
Abstract
Objective. To evaluate the rate of somatic NLRP3 mosaicism in an Italian cohort of mutation-negative patients with cryopyrin-associated periodic syndrome (CAPS).
Methods. The study enrolled 14 patients with a clinical phenotype consistent with CAPS in whom Sanger sequencing of the NLRP3 gene yielded negative results. Patients’ DNA were subjected to amplicon-based NLRP3 deep sequencing.
Results. Low-level somatic NLRP3 mosaicism has been detected in 4 patients, 3 affected with chronic infantile neurological cutaneous and articular syndrome and 1 with Muckle-Wells syndrome. Identified nucleotide substitutions encode for 4 different amino acid exchanges, with 2 of them being novel (p.Y563C and p.G564S). In vitro functional studies confirmed the deleterious behavior of the 4 somatic NLRP3 mutations. Among the different neurological manifestations detected, 1 patient displayed mild loss of white matter volume on brain magnetic resonance imaging.
Conclusion. The allele frequency of somatic NLRP3 mutations occurs generally under 15%, considered the threshold of detectability using the Sanger method of DNA sequencing. Consequently, routine genetic diagnostic of CAPS should be currently performed by next-generation techniques ensuring high coverage to identify also low-level mosaicism, whose actual frequency is yet unknown and probably underestimated.
- GENETIC STUDIES
- PEDIATRIC RHEUMATIC DISEASES
- INFLAMMATION
- NEUROLOGIC MANIFESTATIONS
Cryopyrin-associated periodic syndrome (CAPS) is a group of monogenic autoinflammatory diseases that includes familial cold autoinflammatory syndrome (FCAS), Muckle-Wells syndrome (MWS), and chronic infantile neurological cutaneous and articular syndrome (CINCA). These phenotypes are characterized by chronic or recurrent urticaria-like rash associated with a number of systemic symptoms, and potential structural organ damage. CAPS phenotypes are caused by de novo or dominant mutations of the NLRP3 gene1, which encodes cryopyrin, a key protein of the multiprotein complex called inflammasome that generates the active form of caspase-1 and interleukin 1β (IL-1β)2. Among CAPS patients initially described as NLRP3 mutation-negative, next-generation sequencing (NGS) techniques revealed somatic mutations in this gene in 35% of CINCA cases3,4 and 12.5% of MWS5. To date, the clinical phenotype of patients carrying somatic NLRP3 mosaicism has been indistinguishable from that of patients with germline mutations, with the exception of milder neurological signs3,4,6,7,8.
In patients with CINCA, 2 different reports demonstrated that somatic NLRP3 mosaicism occurs at comparable frequency in cells from both myeloid and lymphoid lineages as well as from cells of ectodermic derivation4,8. More recently, myeloid-restricted somatic NLRP3 mutations have been described in 2 patients with late-onset but otherwise typical CAPS9,10, and in 2 adult patients with the variant-type of Schnitzler syndrome11.
In our current study, we have collected Italian patients presenting with the 3 CAPS phenotypes that were NLRP3-negative in studies using the Sanger method of DNA sequencing, with the aim of evaluating the rate of somatic NLRP3 mosaicism and the involved cellular lineages.
MATERIALS AND METHODS
Patients
The present study enrolled 14 patients affected with clinical symptoms attributable to a CAPS phenotype and NLRP3-negative in genetic studies based on the Sanger method of DNA sequencing. Except for Patient 11, the patients satisfied diagnostic criteria by both Federici, et al12 and Kuemmerle-Deschner, et al13. Patient 11 was included in the study for the presence of repeated episodes of cold-induced fever attacks with elevation of acute-phase reactants. Genomic DNA samples have been extracted from both whole blood and different cellular types by the salting-out method. The study was approved by the Gaslini Ethics Board on May 6, 2004, and was conducted in accordance with the Declaration of Helsinki.
Molecular genetics
For amplicon-based deep sequencing (ADS) studies, amplicons covering all exons of the NLRP3 gene were generated by PCR designed in-house. Library preparation, control quality, and quantification were performed according to manufacturers’ instructions. Emulsion PCR was performed on a One Touch2 platform, and sequencing was performed on an Ion Torrent PGM platform using the Ion Torrent PGM HiQ Sequencing kit. The obtained sequences were analyzed using the Torrent Server and the Ion Reporter softwares (Thermo Fischer Scientific Inc.).
Functional analysis of NLRP3 mutations
The functional consequences of the identified NLRP3 variants were evaluated by means of 2 already-described alternative in vitro assays7. Briefly, the capability of wild type (wt) and mutant NLRP3 to induce necrosis-like cell death in the THP1 cell line has been quantified by FACS after staining with 7-Amino-actinomycin D. Otherwise, dual-luciferase reporter assay in HEK293FT cells was applied to evaluate induction of nuclear factor-κB (NF-κB) activity after transfection with wt and mutant NLRP3 in the presence or absence of its partner ASC (PYCARD).
Purification of immune cell subsets from peripheral blood
Different subpopulations of peripheral blood from patients 1, 6, and 7 were separated by magnetic microbeads CD66b+ (neutrophils) followed by staining with monoclonal antibodies (mAb) anti-CD19 (B cells), CD3 (T cells), CD16, and CD56 (NK cells), CD14 (monocytes; Beckton Dickinson or Biolegend), and cell sorter technology (FACS ARIA). In Patient 2, CD66b+ neutrophils, CD14+ monocytes, and CD19+/CD3+/CD56+ total lymphocytes were separated from whole blood through cell sorting. Subsequently, B, T, and NK cells were isolated after Ficoll separation of peripheral blood mono-nuclear cells, staining with above mAb, and sorting.
RESULTS
The main clinical characteristics of the enrolled patients are summarized in Table 1 and Supplementary Table 1 (available with the online version of this article). ADS revealed somatic NLRP3 mosaicism in unfractionated peripheral blood in 4 patients (Table 2). The 3 patients with CINCA syndrome (pts 2, 6, and 7) turned out to be carriers of a somatic NLRP3 mosaicism. The amino acid substitutions p.Y563C and p.G564S have never been identified in more than 60,000 human exomes14, while the variant p.T433I has been reported as a disease-causing mutation in mosaic status4. Interestingly, Patient 2 displayed a clear neurological involvement, with severe and persistent headache, bilateral papilledema, sensorineural deafness, and progressive cognitive impairment. This last condition is to date very rare in the case of somatic mutations4 (Table 2). In addition, brain magnetic resonance imaging (MRI), performed on a patient aged 37 years, showed mild loss of white matter volume with enlargement of the lateral ventricles, associated with non-specific periventricular white matter lesions and slightly prominent cerebral sulci (Figure 1).

Brain magnetic resonance imaging performed on a 37-year-old patient. Axial fluid attenuated inversion recovery (A) and T2-weighted (B) images reveal mild loss of white matter volume with secondary ventricular enlargement. A few nonspecific hyperintense lesions are noted in the periventricular regions (arrows). Coronal T2-weighted (C) and sagittal T1-weighted (D) images reveal slightly prominent cerebral sulci.
Features of patients with CAPS analyzed for NLRP3 mosaicism. All patients are characterized by increment of inflammatory markers at clinical diagnosis. No patients showed AA amyloidosis.
Frequency of identified somatic mutation of NLRP3. Each frequency value corresponds to the average of 3 independent experiments.
A somatic NLRP3 mutation was detected in only 1 out of 9 patients with MWS (11.1%). This patient carried the missense p.R260P amino acid substitution, which has been reported as a disease-causing mutation in germline status15. By contrast, neither of the 2 patients with FCAS turned out to be a carrier of a somatic NLRP3 mutation.
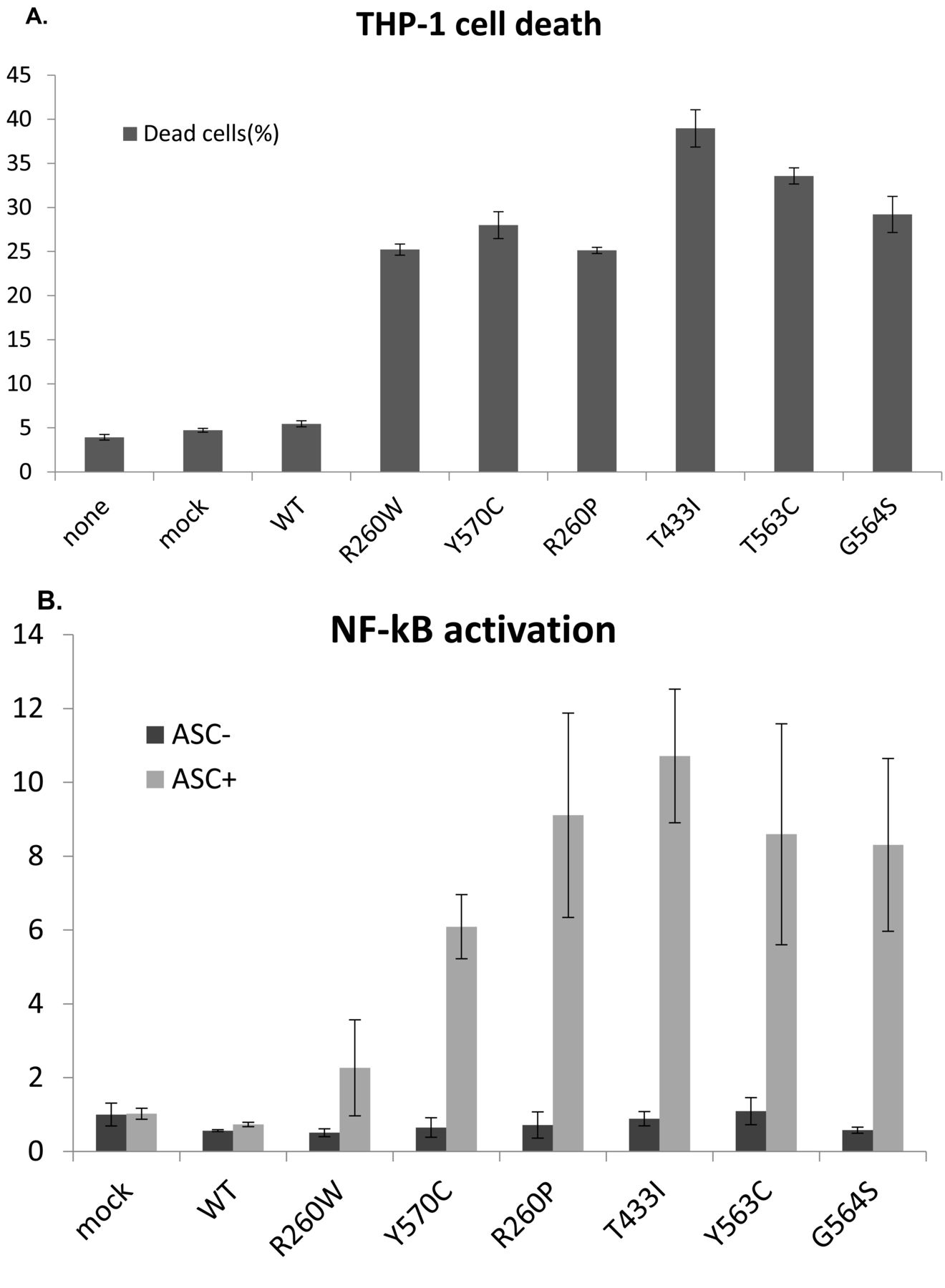
To confirm the functional role of the detected somatic mutations, we have taken advantage of 2 different in vitro assays. Compared to wt cryopyrin, mutant forms of protein rapidly induce necrotic cell death when transiently transfected in human monocytic THP-1 cells (Figure 2A). In addition, co-expression of mutant cryopyrin with its partner ASC in HEK293FT cells causes enhanced NF-κB reporter activity (Figure 2B). Notably in both analyses, the effect of the newly identified mutations appears to be stronger than that of the known disease-causing mutations p.R260W and p.Y570C.
{kind=link}
{kind=link}
Functional analysis of NLRP3 mutations. (A) Mutated NLRP3 proteins induce cell death in transfected THP-1 cells. Bars represent percentages of dead cells (7-Amino-actinomycin D–positive) among the NLRP3-GFP+ cells as means of triplicate experiments. Error bars correspond to SD. Constructs with NLRP3-GFP–bearing p.Arg260Trp and p.Tyr570Cys (R260W and Y260C) are used as positive controls. Activity of wt NLRP3-GFP is represented. None = not transfected; mock = cells subjected to transfection protocol without vectors. (B) Mutated NLRP3 induce NF-κB activation in transfected HEK293FT cells when ASC is cotransfected to reconstitute an active inflammasome. NF-κB activation is shown as fold-change compared to the activity of empty control vector. Bars represent means of triplicate experiments and error bars correspond to SD. ASC+ = transfection of NLRP3 expression vector together with ASC expression vector; ASC− = transfection of NLRP3 expression vector with empty expression vector. WT: wild type; NF-κB: nuclear factor-κB.
As previously reported, the frequency of the mutated NLRP3 allele remained similar in blood samples obtained over several years5.
In some patients, the functional analysis of IL-1 secretion was performed with variable results. Patients 6 and 7 displayed a high IL-1 secretion after lipopolysaccharide (LPS) stimulation similar to that observed in patients with CAPS carrying germline mutations (data not shown). Patient 2 displayed a slight increase of IL-1 secretion after LPS stimulation with the same accelerated pattern observed in CAPS16 (Supplementary Figure 1, available with the online version of this article).
In all patients bearing somatic NLRP3 mosaicism, we have analyzed the presence of the mutant allele in different tissues. All blood cell subpopulations as well as the epithelial cells displayed the mutant allele. Apart from a few exceptions, no significant differences in the frequency of mutant allele were observed. In particular, 2 of 5 ectodermic tissues (epithelial cells from urine and hair bulb) were negative for mutant allele in Patient 2, and monocytes displayed a very low frequency in Patient 1.
DISCUSSION
In recent years, a number of studies have demonstrated that somatic NLRP3 mutations are responsible for CAPS phenotypes in a relevant percentage of patients who were negative in previous studies using conventional Sanger sequencing. Actually, this phenomenon accounts for 35% of CINCA and 12.5% of MWS patients3,4,5. To date, no evidence of somatic NLRP3 mosaicism has been reported in FCAS. Our results in Italian patients confirm that somatic NLRP3 mosaicism is a genetic mechanism underlying CINCA and MWS phenotypes.
In line with published data, 50% of the NLRP3 mutations detected in this work are novel, and one of the remaining variants had been described only in mosaic status4. This observation enforces the hypothesis that these mutations in germline status could be incompatible with life. Actually, in vitro functional studies clearly showed their gain-of-function effect, even stronger for mutations found in mosaic status compared to germline mutations.
To date, the patients carrying somatic NLRP3 mosaicism did not present clear clinical differences when compared to patients carrying germline mutations, with the exception of milder neurological signs in patients with mosaicism4. To our knowledge, Patient 2 represents the first patient with CINCA carrying a somatic NLRP3 mutation with minor structural brain abnormalities on MRI, including reduction of white matter volume and prominent cerebral sulci. The amino acid exchange he carries (p.G564S) is novel, suggesting that this neurological implication could be a peculiar phenotypic effect, or alternatively could be related to the specific tissue distribution of the mutant allele, which is not easily analyzable.
Similar considerations could also explain a severe phenotype in case of very low mosaicism frequency, as in Patient 6 (2.7% of mutated allele), characterized by early onset, arthritis, and bone dysplasia. Notably, because a given frequency of the mutated allele corresponds to a double frequency of heterozygous cells, the presence of an active NLRP3 mutation in only a portion of about 5.4% of circulating leukocytes appears to be sufficient to cause the clinical manifestations of CINCA syndrome. But a lower frequency (2.4%) of a different mutation (p.T348M) was not sufficient to induce the clinical manifestations in the asymptomatic mother of a patient with MWS17, thus confirming that not only the amount of mutant allele but also the characteristic of the mutation and likely its tissue distribution are going to determine the phenotypic effects of an NLRP3 mosaicism.
The patients here detected as carrying somatic NLRP3 mosaicism display the variant allele in tissues derived from different embryonic layers, suggesting that the somatic mutational event occurred very early during the ontogenesis.
Consistent with most of the patients already described3,4,5,7, the level of mosaicism in our cases is below 15%, commonly considered the threshold of detectability using conventional Sanger sequencing. In contrast with some other countries, in Italy the lack of a positive genetic test at standard Sanger analysis does not prevent a CAPS diagnosis in patients with a clear phenotype. This leads to the use of anti–IL-1 treatments, as experienced by our patients, with the same excellent results observed in patients with germline mutation18. However, a correct genetic diagnosis is essential to allow the patient to make informed choices about his/her reproductive future. Though the somatic mosaicism rate may differ in tissues and it is not usually determined in gonadal tissues, the risk of transmission of the mutated allele to the offspring is regarded as likely low, and the genetic counseling is quite challenging. Indeed, there is evidence that somatic NLRP3 mutations also affect germline cells, with consequent vertical transmission17.
Detection of mosaicism in autoinflammatory disease is increasing, having recently been demonstrated also in the TMEM173, NOD2, TNFRSF1A, and NLRC4 genes19,20,21,22. To date, among CAPS-like phenotypes, genetic heterogeneity has been found in a small proportion of FCAS cases23,24 and in a single patient with CINCA22, while an approach based on whole exome sequencing to search for a novel candidate gene was able to identify only a mutation of the NLRP3 gene in mosaic status25. For this reason, in the case of clinical features attributable to CAPS, we strongly support the need to replace Sanger sequencing with NGS techniques that ensure high coverage to identify low-level mosaicism, whose actual frequency is still unknown and likely underestimated.
Our work confirms the presence of somatic NLRP3 mosaicisms in Italian patients with CAPS who were negative for germline mutations with conventional Sanger analysis. This evidence highlights the need to update the routine molecular diagnosis for CAPS by means of high-coverage NGS, which can also identify low-level mosaicism in the NLRP3 gene.
ONLINE SUPPLEMENT
Supplementary material accompanies the online version of this article.
Acknowledgment
We are extremely grateful to all the families who have participated in this study, and to Loredana Velo and Laura Carenini for excellent secretarial assistance.
Footnotes
Supported by Giovani Ricercatori–Ricerca Finalizzata grant no. GR-2010-2315933 to SB, and partially funded by the Italian Ministry of Health through both “Cinque per mille” and Ricerca Corrente to the Gaslini Institute, Ricerca Telethon GGP14144 to MG and CERCA Programme/Generalitat de Catalunya (JIA), SAF2015-68472-C2-1-R grant from the Spanish Ministry of Economy and Competitiveness and Fondo Europeo de Desarrollo Regional (JIA) and AC15/00027 grant from the Instituto de Salud Carlos III/Transnational Research Projects on Rare Diseases (JIA).
- Accepted for publication June 30, 2017.
REFERENCES
- 1.
- 2.
- 3.
- 4.
- 5.
- 6.
- 7.
- 8.
- 9.
- 10.
- 11.
- 12.
- 13.
- 14.
- 15.
- 16.
- 17.
- 18.
- 19.
- 20.
- 21.
- 22.
- 23.
- 24.
- 25.