
To the Editor:
In May 2010, a 36-year-old man with a history of active terminal ileal Crohn’s disease presented to the emergency department with a 3-week history of proximal lower extremity muscle weakness. Six months previously (October 2009–April 2010), the Crohn’s disease was initially treated with multiple courses of prednisone and azathioprine 250 mg daily. Despite this regimen, he continued to have extraintestinal manifestations of Crohn’s disease, including morning stiffness, polyarthralgias of the hands and feet, early satiety, and a 30-lb weight loss in 4 months. Adalimumab was prescribed for management of his Crohn’s symptoms.
Prior to administration of adalimumab, laboratory investigations in March 2010 revealed erythrocyte sedimentation rate (ESR) 107 mm/h; antinuclear antibody (ANA) 1:640 in a positive homogenous pattern; positive anti-U1 RNP and negative anti-SSA/Ro, anti-SM, anti-SSB/La, anti-SCl-70, anti-Jo-1, and native DNA antibodies; rheumatoid factor (RF) 17 KIU/ml (normal 0–19); antibodies to cyclic citrullinated peptide (anti-CCP) < 8 U/ml; C3 1.46 g/l (normal 0.90–1.80), and C4 0.18 g/l (normal 0.10–0.40). Creatine kinase (CK) was normal at 118 U/l (normal 55–197).
On April 20, 2010, he received an induction dose of adalimumab 160 mg subcutaneously. On May 3, he developed sudden onset of symmetrical edema of his face and neck. On May 4, he received a second 80 mg dose of adalimumab; at Week 4, a maintenance dose of 40 mg was given. His facial edema worsened after this last dose and he experienced night sweats and diarrhea. He had increasing proximal muscle weakness and myalgias of the lower extremities that interfered with mobility. He also had morning stiffness and joint swelling of his knees, ankles, wrists, and metacarpals bilaterally. At this time, his only medication was adalimumab.
Repeat laboratory investigations while on adalimumab included elevated C-reactive protein (CRP) 106 mg/l and ESR 60 mm/h, RF 17 KIU/ml, anti-CCP < 8 U/ml, ANA 1:640 in a homogenous pattern, positive RNP, and borderline positive Sm antibodies. The other autoantibodies remained negative; native DNA antibodies were negative. C3 level was 0.69 g/l and C4 was 0.11 g/l. CK was now elevated to 3709 U/l (Figure 1). Electromyography studies showed proximal muscle inflammation consistent with dermatomyositis (DM), later proven with a muscle biopsy. Left deltoid muscle biopsy confirmed necrosis, macrophages (CD68), and leukocyte common antigen-positive lymphocytes. C1 esterase inhibitor deficiency was excluded when serology was negative. Anti-adalimumab antibodies were positive.

Relationship of creatine kinase with the onset of adalimumab treatment (start date April 20, 2010) and infliximab treatment (start date August 10, 2011). Bottom arrows: prednisone 30 mg.
Edema was most likely secondary to use of adalimumab. This was thought to be a serum sickness or allergic reaction to adalimumab. Adalimumab was discontinued after the May 4, 2010, dose and the edema of the face and neck resolved over the next 3 weeks. The fatigue, weight loss, night sweats, and myalgias continued. In May 2010, methotrexate (MTX) 10 mg weekly and prednisone 30 mg daily were prescribed for active myositis. The morning stiffness, myalgias, and polyarthralgias improved after the first week on prednisone. Taking MTX and prednisone, he continued to have bowel symptoms.
Due to intolerance to adalimumab and prednisone-resistant Crohn’s disease, the patient was started on infliximab 5 mg/kg on August 10, 2010. He received a second dose 2 weeks later (5 mg/kg) and his third and final dose (5 mg/kg) on September 15, 2010, before presenting to hospital. He was concurrently on a month of prednisone 30 mg daily. He was also taking MTX 10 mg weekly. Early in September 2010, he had developed what he described as lesions resembling a heliotrope rash and Gottron’s papules that self-resolved. This was not observed by us. CK on September 4 was 1222 U/l. Due to worsening myalgias and rising CK, the prednisone and MTX were increased from 30 mg to 50 mg and from 10 mg to 20 mg weekly, respectively, in mid-September.
In October 2010, he developed severe bilateral (left worse than right) lower extremity mid-thigh to foot myalgias, erythema, and edema, leading to inability to ambulate due to pain, and this led to another hospital admission. He denied systemic or gastrointestinal symptoms. Other current medications, along with infliximab, included prednisone 50 mg daily and MTX 20 mg orally once weekly.
On examination, he was afebrile and hemodynamically stable. Bilateral lower extremities (left more than right) were red, swollen, and markedly tender to touch, especially over the gastrocnemius and hamstring muscles. Ranges of motion of the hip, knee, and ankle joints and neurovascular examination were intact. There was no synovitis. Although muscle strength testing was limited by pain, strength was 5/5 in both upper and lower extremities. There were no rashes of DM.
Repeat laboratory investigations (October 2010) after the start of infliximab were as follows: ESR 101 mm/h, CRP 40 mg/l, RF 17 KIU/ml, and anti-CCP < 8 U/ml. Serum autoantibodies remained unchanged: positive anti-U1-RNP and borderline positive anti-Sm and the others including anti-SSA/Ro, anti-SSB/La, anti-SCl-70, anti-Jo-1, and native DNA antibodies remained negative. CK was 183 U/l.
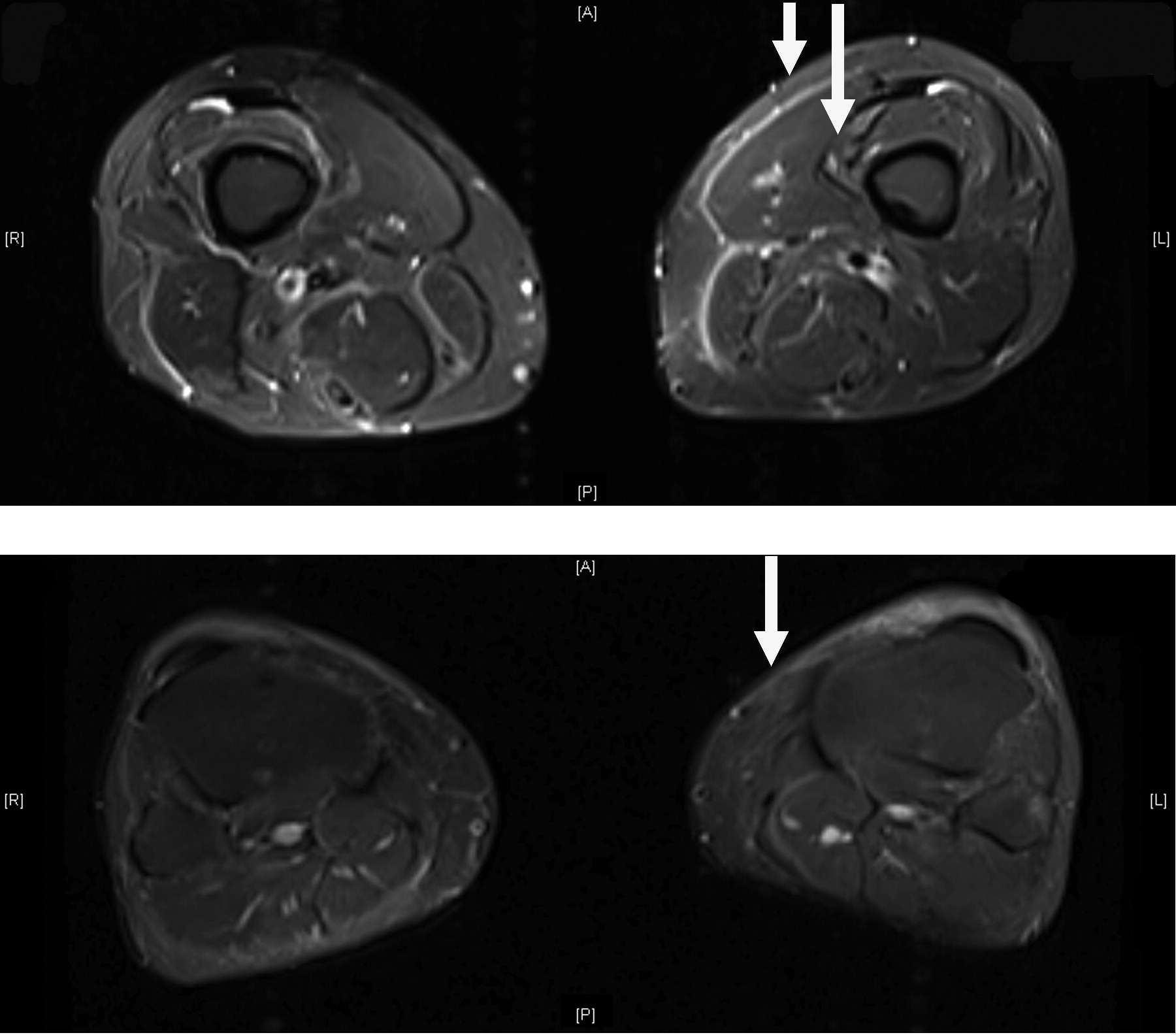
It was puzzling that there was severe myalgia despite the now normal CK. Radiographs of the ankle mortise and foot were normal. Magnetic resonance imaging (MRI) of the lower extremity revealed fascial fluid in posterior compartments of both thighs. There was also some mild increased signal involving the hamstring musculature bilaterally, most prominently involving the distal portion of the biceps femoris (greater on the left). There was also some increased signal within the medial and lateral head of the gastrocnemius muscle in the left calf. Edema and inflammation were present in skin, subcutaneous tissue, and fascia (Figure 2). This was nonspecific but most likely related to diffuse myositis with bilateral diffuse fasciitis. The differential diagnosis of muscle tenderness in our patient taking high doses of immunosuppressants included relapse of DM, pyomyositis, necrotizing fasciitis, autoimmune myositis, and drug-induced myositis.
{kind=link}
{kind=link}
Top panel: Magnetic resonance imaging (MRI) scan October 2010 shows inflammation in skin, subcutaneous tissue, and fascia (short arrow) of posterior compartments of thighs, greater on left side (image right). Long arrow indicates normal muscle. Bottom panel: March 2011; MRI resolution of inflammation (arrow) in posterior compartments of thigh after stopping infliximab.
Fasciitis is, surprisingly, a common early lesion of DM1. Fasciitis can progress into DM. Yoshida, et al1 studied whether fasciitis exists histologically after onset on DM muscle symptoms by performing an en bloc biopsy of muscles; this revealed abnormal hyperintense areas on MRI. Biopsies involving skin, subcutaneous tissues, fascia, and muscle were taken from patients with newly diagnosed DM. These patients had not received immunosuppressives and/or had taken < 10 mg prednisone. The biopsy results showed inflammatory infiltrates around fascial small blood vessels in all 14 patients with early DM. It is concluded that fasciitis is one of the early pathological lesions in DM and is one cause of muscle symptoms in DM1. Perivascular inflammatory cell infiltration occurs originally in the fascia and expands into muscle along fascia and interfascicular septum1. As in our case, edema and inflammation were present on MRI in skin, subcutaneous tissue, and fascia soon after the relapse of DM.
Cutaneous involvement of DM varies. Patients may present with heliotrope rash or Gottron’s lesions. Facial swelling may be a first manifestation of DM2. Our patient’s edema of the face and neck in May 2010 was most likely a cutaneous feature of DM.
The causality between DM and anti-tumor necrosis factor-α (anti-TNF-α) agents can be clearly described in our case. Prior to starting anti-TNF-α therapies, he had positive ANA and anti-U1-RNP antibodies. Adalimumab and infliximab worsened the clinical picture in a patient with evidence of an autoimmune condition. DM developed while he was receiving uninterrupted adalimumab therapy. Removal of adalimumab improved the myalgias within 3 weeks and normalized the CK within 6 weeks. Prednisone and MTX were effective for his myositis. He continued to have incapacitating Crohn’s symptoms and therefore was prescribed a second anti-TNF-α agent, infliximab. Infliximab could be considered in this case the “re-challenge agent,” leading to severe DM with fasciitis once again, despite high-dose prednisone 50 mg and weekly oral MTX 20 mg. The fasciitis was likely a lesion within the spectrum of DM. Stopping the infliximab resolved his symptoms and the fasciitis within 3 weeks. This was proven by a repeat MRI (March 2011), which demonstrated resolution of edema and inflammation (Figure 2). The prompt resolution is highly suggestive of an anti-TNF inhibitor-induced autoimmune syndrome consisting of DM with fasciitis.
Cases of myopathies associated with anti-TNF inhibitors have been described. There are 5 cases of infliximab or etanercept-induced anti-Jo antibody-positive polymyositis3, 1 case of etanercept-induced DM, 1 case of axonal neuropathy associated with adalimumab, 1 case of interstitial myositis with demyelinating neuropathy associated with infliximab, and 1 case of necrotizing myositis associated with etanercept4,5,6,7. There are 5 other cases of DM treated with etanercept; these patients had worsening of their muscle weakness and no improvement of the rash8.
No cases have been described in which fasciitis was the predominant clinical presentation.
Anti-TNF inhibitors are increasingly being associated with autoimmune syndromes6,8,9. As in our case and others, practitioners should be aware of this rare association between anti TNF-α inhibitors and DM. Fasciitis may be the initial presenting feature of DM. If patients develop myositis while receiving anti-TNF therapy, the causality of such therapy needs to be considered.
REFERENCES
- 1.
- 2.
- 3.
- 4.
- 5.
- 6.
- 7.
- 8.
- 9.