


Abstract
The concept that the immune system can communicate with peripheral sensory neurons to modulate pain is based mostly on documented interactions between opioid ligands and receptors. Such findings may have broad implications for the development of safer pain medication. Innovative strategies take into account that analgesics should be particularly active in pathological states rather than producing a general suppression of the central nervous system, as with conventional morphine- or cannabinoid-like drugs. Inflammation of peripheral tissue leads to increased functionality of opioid receptors on peripheral sensory neurons and to local production of endogenous opioid peptides. In addition, endocannabinoids were detected in leukocytes, but their role in pain modulation has yet to be addressed. Future aims include the development of peripherally restricted opioid agonists, selective targeting of opioid-containing immune cells to sites of painful injury, and the augmentation of peripheral ligand and receptor synthesis (e.g., by gene therapy). Similar approaches may be pursued for cannabinoids. The ultimate goal is to avoid detrimental side effects of currently available analgesics such as respiratory depression, cognitive impairment, addiction, gastrointestinal bleeding, and thromboembolic complications.
I. Introduction
Inflammation is an essential component of a large group of painful syndromes, including arthritis, inflammatory back pain (e.g., spondyloarthritis), neurogenic migraine, inflammatory lesions of the central and peripheral nervous system (neuropathic pain), cancer pain, disorders of the immune system, burns, wounds, traumatic and postoperative pain (for review, see Stein et al., 2009). Within peripheral damaged tissue (e.g., skin, muscles, joints, viscera), primary afferent neurons transduce noxious mechanical, chemical, or thermal stimuli into action potentials. The cell bodies of these neurons are located in the trigeminal and dorsal root ganglia (DRG1) and give rise to myelinated (Aδ) and small-diameter unmyelinated axons (C fibers). Most of the latter are sensitive to capsaicin, a ligand at the transient receptor potential vanilloid-1 (TRPV1) channel. C fibers are considered the dominant fibers in clinical pain and have thus been coined “nociceptors. ” After modulation within the primary afferent neurons and spinal cord, nociceptive signals reach the brain, where they are finally perceived as “pain,” within the context of cognitive and environmental factors (Woolf and Salter, 2000). Primary afferent neurons are of particular interest from a therapeutic perspective because they are the initial generators of nociceptive impulses. Thus, if one finds ways to inhibit the sensitization and/or excitation of these neurons, subsequent central events such as wind-up, sensitization, and plasticity may be prevented.
Attention has traditionally been focused on the characterization of proinflammatory and proalgesic effects elicited by the myriad of mediators occurring in injured tissue (e.g., cytokines, neuropeptides, kinins, prostaglandins). Concurrently, however, endogenous mechanisms counteracting pain and inflammation are mounted. In the periphery, such effects are produced by interactions between leukocyte-derived opioid peptides and opioid receptors on peripheral endings of primary afferent neurons and by anti-inflammatory cytokines (Stein et al., 2003; Rittner et al., 2008). These findings have opened new perspectives for the treatment of pain, particularly regarding the use of opioids. Conventional opioids are the most powerful drugs to alleviate severe pain, but their use is hampered by side effects such as depression of breathing, nausea, clouding of consciousness, constipation, addiction, and tolerance (Table 1) (Zöllner and Stein, 2007). Thus, the development of drugs lacking such effects has always been a major goal in pain research. The discovery of opioid and cannabinoid (CB) receptors on sensory nerves has now put this goal within reach. Subsequent to studies on the local application of conventional opioids in peripheral damaged tissue, a new generation of opioid drugs and formulations unable to pass the blood-brain barrier, thereby avoiding central adverse effects, is now emerging. This review will focus on the localization, trafficking, and function of peripheral opioid receptors, the production and release of opioid peptides in peripheral inflamed tissue, implications for analgesia and opioid tolerance, therapeutic issues in inflammatory diseases, and knowledge gaps defining future research directions. Because there is no evidence on the involvement of leukocyte-derived endocannabinoids in pain modulation, and peripheral analgesic effects of exogenously applied cannabinoids have been subject of numerous recent review articles (Akopian et al., 2009; Anand et al., 2009; Kress and Kuner, 2009; Sagar et al., 2009; McDougall, 2011), the cannabinoid system will be addressed only briefly.
Opioid receptors and ligands
II. Opioids
A. Opioid Receptors
1. Opioid Receptor Types, Signal Transduction, and Receptor Trafficking.
Early binding studies and bioassays defined three main types of opioid receptors in the central nervous system, the μ-, δ-, and κ-receptors (Table 1). Additional receptor types were proposed (e.g., σ, ε, orphanin) but are no longer considered “classic” opioid receptors (for review, see Kieffer and Evans, 2009). The identification and sequence analysis of cDNA and the selective deletion of opioid receptor genes in mice confirmed the existence of only three genes and allowed for the study of individual opioid receptor types with regard to their pharmacological profile, intracellular effector coupling, anatomical distribution, and regulation of expression (Kieffer and Evans, 2009). Opioid receptors belong to the family of seven-transmembrane G protein-coupled receptors (GPCRs) and show 50 to 70% homology between their genes. Additional pharmacological subtypes may result from alternative splicing, post-translational modifications, or receptor oligomerization (Waldhoer et al., 2004; for review, see Kieffer and Evans, 2009; van Rijn et al., 2010). Opioid receptors are expressed by central and peripheral neurons, by neuroendocrine (pituitary, adrenals), immune, and ectodermal cells (for review, see Zöllner and Stein, 2007).
The signaling pathways of opioid receptors are well characterized. After ligand binding, conformational changes allow intracellular coupling of heterotrimeric Gi/o proteins to the C terminus of the receptor. At the Gα subunit, GTP replaces GDP and dissociation of the trimeric G protein complex into Gα and Gβγ subunits ensues. Subsequently, these subunits can inhibit adenylyl cyclases and thereby inhibit cAMP production and/or directly interact with different ion channels in the membrane (Fig. 1). Ion channels are mainly regulated by direct interaction with Gβγ subunits (Herlitze et al., 1996; Tedford and Zamponi, 2006; Lüscher and Slesinger, 2010). All three opioid receptors can modulate pre- and postsynaptic N-, T-, and P/Q-type Ca2+ channels, suppress Ca2+ influx, and thereby attenuate the excitability of neurons and/or reduce neurotransmitter release. A prominent example is the inhibition of substance P and calcitonin gene-related peptide (CGRP) release (both are pronociceptive and proinflammatory neuropeptides) from central and peripheral terminals of primary afferent neurons (Fig. 1) (Yaksh, 1988; Khasabova et al., 2004; Kondo et al., 2005). At the postsynaptic membrane, opioid receptors mediate hyperpolarization by opening G protein-coupled inwardly rectifying K+ (GIRK) channels, thereby preventing neuronal excitation and/or propagation of action potentials (Zöllner and Stein, 2007; for review, see Lüscher and Slesinger, 2010). Various enzymes, such as protein kinases (A and C) and GPCR kinases, can phosphorylate opioid receptors, leading to an increased affinity for intracellular arrestin molecules. The formation of arrestin-opioid receptor complexes leads to opioid receptor desensitization by preventing G protein coupling and promotes internalization via clathrin-dependent pathways. Recycling of opioid receptors and their reintegration into the plasma membrane resensitizes signal transduction, whereas targeting to lysosomes leads to receptor degradation (Fig. 1). GPCR-associated sorting proteins modulate lysosomal sorting and functional down-regulation (Waldhoer et al., 2004; Moore et al., 2007). Additional opioid-modulated pathways can involve N-methyl-d-aspartate receptors, mitogen-activated protein kinase, and phospholipase C (Zöllner and Stein, 2007).
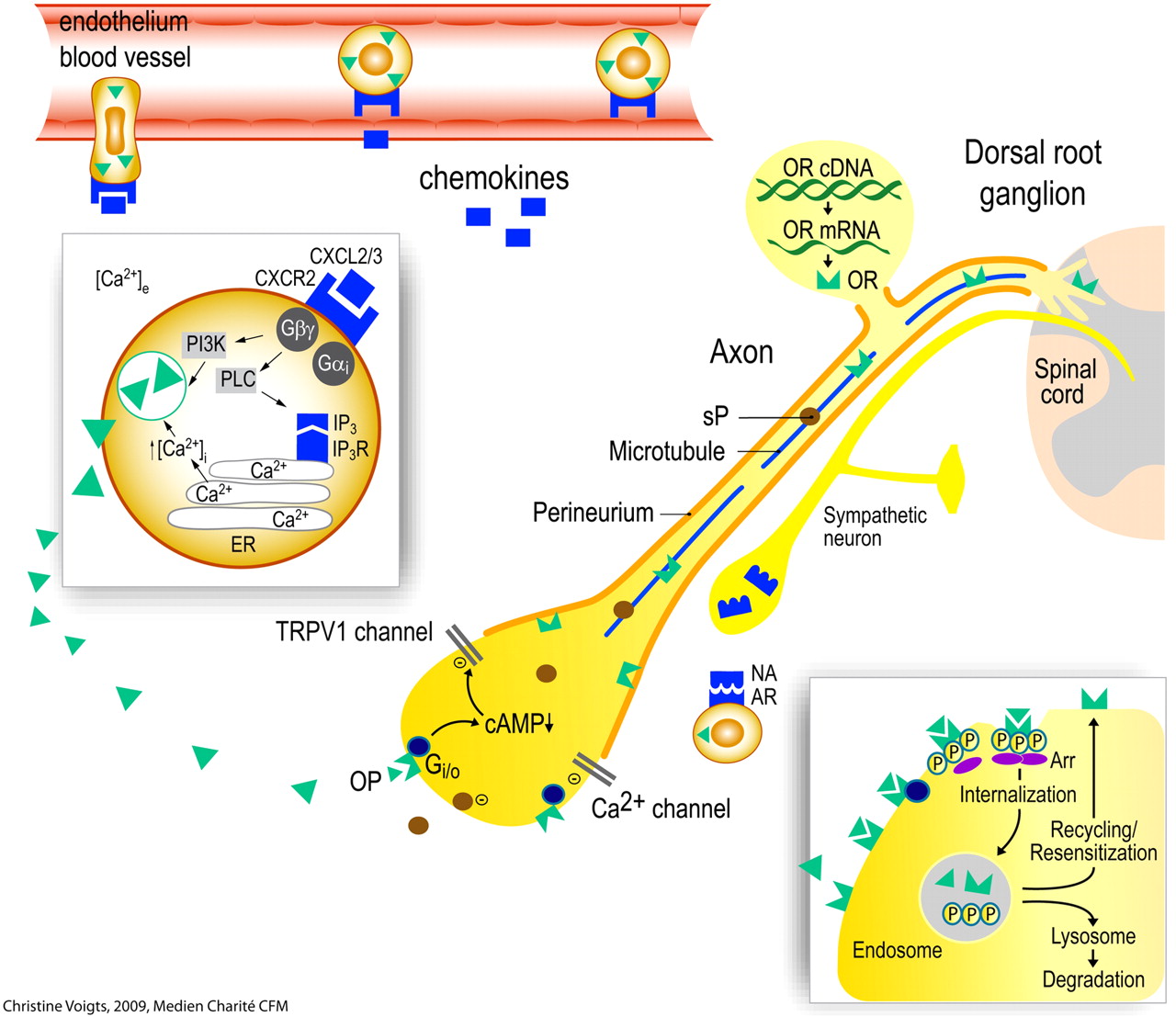
Opioid peptide (OP)-containing leukocytes migrate into inflamed tissue. OP release from polymorphonuclear granulocytes is triggered by. e.g., the chemokine CXCL2/3 (insert). This requires elevation of intracellular Ca2+ through efflux from the endoplasmic reticulum (ER) and (in part) phosphoinositol-3-kinase (PI3K). OP can also be released by noradrenaline (NA; derived from sympathetic neurons) activating adrenergic receptors (AR). Opioid receptors (OR) and neuropeptides [substance P (sP)] are synthesized in the dorsal root ganglion and transported along intra-axonal microtubules into central and peripheral processes of the primary afferent neuron. The permeability of the perineurium is increased in inflamed tissue. Upon activation by endogenous (OP) or exogenous opioids, OR couple to inhibitory G-proteins (Gi/o). This leads to decreased cAMP and inhibition (−) of Ca2+ and/or TRPV1 currents, resulting in reduced excitability of the neuron and in attenuated sP release. Activation of OR stimulates receptor phosphorylation, arrestin (Arr) binding, and internalization (insert). Subsequently, OR can either be degraded in lysosomes or dephosphorylated and recycled to the cell membrane. Recycling counteracts the development of tolerance to opioid agonists. cAMP, cyclic adenosine monophosphate; IP3, inositol 1,4,5-triphosphate; PLC, phospholipase C. [Adapted from Busch-Dienstfertig M and Stein C (2010) Opioid receptors and opioid peptide-producing leukocytes in inflammatory pain—basic and therapeutic aspects. Brain Behav Immun 24:683–694. Copyright © 2010 Elsevier. Used with permission.]
2. Opioid Receptors on Peripheral Sensory Neurons.
Peripheral antinociceptive effects of opioids were approximately 30 years ago (for review, see Stein, 1993). In the early 1990s, evidence began to accumulate that such effects are mediated by opioid receptors localized on peripheral sensory neurons (Barthó et al., 1990; Stein et al., 1990b; Stein, 1995). Since then, many studies have shown that opioid receptors are expressed in small-, medium-, and large-diameter DRG neurons, together with prototypical sensory neuropeptides such as substance P and CGRP (for review, see Busch-Dienstfertig and Stein, 2010). A recent debate has evolved around the question whether μ- and δ-opioid receptors are coexpressed with each other and with the above-mentioned neuropeptides and whether these receptors modulate distinct nociceptive stimuli (Scherrer et al., 2009; Wang et al., 2010). However, it is important to keep in mind that these studies were carried out in animals without painful tissue injury and that various types of damage (e.g., inflammation, nerve compression) can dramatically change gene and protein expression in DRG neurons (see section II.A.5). Opioid receptors are transported to the peripheral nerve terminals (Hassan et al., 1993; Li et al., 1996; Puehler et al., 2004; Mousa et al., 2007a) and couple to Gi/o proteins that inhibit adenylyl cyclases and modulate ion channels (Zöllner et al., 2003; Zöllner et al., 2008). The inhibition of Ca2+ channels seems to be a major mechanism for the opioid-induced suppression of DRG neuron functions (Akins and McCleskey, 1993; Acosta and López, 1999; Khasabova et al., 2004; Wang et al., 2010). Interactions between different opioid receptor types, possibly via heterodimerization, can facilitate coupling to Ca2+ channels (Walwyn et al., 2005, 2009). Activation of peripheral opioid receptors also suppresses tetrodotoxin-resistant Na+ channels (Gold and Levine, 1996), nonselective cation currents (Ingram and Williams, 1994), purinergic 2X receptor-mediated currents (Chizhmakov et al., 2005), as well as TRPV1 currents via Gi/o and the cAMP/protein kinase A pathway (Chizhmakov et al., 2005; Endres-Becker et al., 2007). Colocalization of GIRK channels and μ-opioid receptors was shown on sensory nerve endings in the epidermis (Khodorova et al., 2003), but no direct evidence of functional coupling or modulation of GIRK channels in DRG neurons has been provided so far. However, morphine was suggested to hyperpolarize DRG neurons by activation of KATP currents via the nitric oxide pathway (Cunha et al., 2010). Furthermore, μ-opioid receptor-mediated hyperpolarization, increased K+ currents, and decreased firing rates of action potentials were shown in neurons from neonatal rats (Takeda et al., 2004). Similar to the central nervous system, β-arrestins regulate the activity and recycling of opioid receptors. Specifically, it was shown that a lack of β-arrestin2 reduced the inhibition of voltage-dependent Ca2+ channels by μ-agonists, diminished constitutive recycling, and increased cell-surface localization of μ-receptors in DRG neurons (Walwyn et al., 2007). In summary, opioid agonists can attenuate the excitability of primary afferent neurons and block the release of proinflammatory neuropeptides (substance P, CGRP) from central and peripheral terminals (Junger et al., 2002; Stein et al., 2003; Khasabova et al., 2004). Particularly within injured tissue, these events led to antinociceptive and anti-inflammatory effects (see sections II.A.5 and II.C).
3. Opioid Receptors on Peripheral Sympathetic Neurons.
Opioid receptor expression in sympathetic postganglionic neurons was also suggested. However, neither opioid receptor mRNA nor protein has been detected in such neurons (Coggeshall et al., 1997; Wenk and Honda, 1999; Ständer et al., 2002; Mousa et al., 2007b). Moreover, whereas the C-fiber neurotoxin capsaicin decreased opioid receptor numbers in the DRG and abolished peripheral opioid antinociception (Barthó et al., 1990; Zhang et al., 1998a; Zhou et al., 1998), chemical sympathectomy with 6-hydroxydopamine did not change the expression of opioid receptors in the DRG or the peripheral analgesic effects of μ-, δ-, and κ-receptor agonists in conditions with or without peripheral inflammation (Zhang et al., 1998a; Zhou et al., 1998).
4. Opioid Receptors on Immune Cells.
Opioid receptors were detected in lymphocytes, monocytes, macrophages, and granulocytes. Leukocytic opioid receptors have similar pharmacological and biochemical characteristics and are encoded by the same genes as neuronal opioid receptors (for review, see Sharp, 2006). Binding of opioid ligands triggers the same signaling pathways as in neuronal cells (e.g., modulation of cAMP, Ca2+, kinases, transcription factors). Opioids were shown to modulate immune cell proliferation, chemotaxis, cytotoxicity, cytokine and chemokine receptor expression, and cytokine synthesis and secretion. These effects were examined predominantly in vitro and were often contradictory, depending on the experimental conditions (e.g., cultured cell types, lines or clones, duration of cultures, doses, and timing of opioid exposure) (for review, see Sacerdote et al., 2003; Sharp, 2006). Thus, leukocytic opioid receptors might contribute to the modulation of inflammation (see section II.C.6). However, their significance in pain transmission has not been directly examined.
5. Plasticity and Regulation of Peripheral Opioid Receptors.
a. Ontogeny of opioid receptor expression in peripheral sensory neurons.
The ontogeny of opioid receptors was examined in the central and peripheral nervous system during preand postnatal development. Using radioligand binding and in situ hybridization techniques, μ-, δ-, and κ-receptor expression was found to be distinct with regard to age, species, tissue, and receptor type. The expression of δ-receptors lags behind that of μ- and κ-receptors in mouse and rat brain (Spain et al., 1985; Zhu et al., 1998). However, in mouse DRG neurons, δ-receptors are the first to be expressed on embryonic day 12.5 (E12.5), followed by the μ- (E13.5) and κ-receptors (E17.5) (Zhu et al., 1998). In rat, a greater proportion of DRG neurons expressing μ- and δ-receptors was found before postnatal day 7 (P7) than at P21 (κ-receptor was not examined) (Beland and Fitzgerald, 2001; Nandi et al., 2004). During the first postnatal week, both opioid receptors were detected in cells of all sizes, but by P21, expression was restricted to small- and medium-diameter cells, suggesting a selective down-regulation in non-nociceptive neurons (Beland and Fitzgerald, 2001; Nandi et al., 2004). This notion was supported by another study examining μ-receptor mRNA and function (Wu et al., 2009). A larger proportion of DRG neurons was consistently responsive to μ-agonists in Ca2+ imaging experiments, and morphine displayed a greater antinociceptive potency to mechanical stimulation in neonatal rats (Nandi et al., 2004). Another study also demonstrated mRNA and functional μ-opioid receptors in rat pups between P6 and P14 (Takeda et al., 2004). In mouse, the postnatal expression of μ-receptors in a subset of DRG neurons seems to be regulated by the transcription factor Runx1 (Chen et al., 2006). There are some phylogenetic differences in opioid receptor expression in the spinal cord and DRG across mammalian species. In monkey and human DRG neurons, δ-receptors are more abundant compared with rodents (Mennicken et al., 2003).
b. Influence of inflammation.
Painful inflammation of peripheral tissue (of varying duration) was most extensively studied as a regulatory stimulus of opioid receptor plasticity in adult sensory neurons. Both the systemic and the local application of μ-, δ-, and κ-receptor agonists elicits significantly more pronounced analgesic effects in injured tissue than in noninjured tissue of animals and humans (for review, see Stein, 1995; Stein et al., 2003). This intriguing finding has stimulated extensive research into the underlying mechanisms.
In cultured trigeminal ganglion neurons, pretreatment with bradykinin was found to stimulate the trafficking of intracellular δ-receptors to the plasma membrane (Patwardhan et al., 2005). Furthermore, priming with bradykinin or a protease-activated receptor agonist led to a more potent inhibition of CGRP release and cAMP accumulation by opioid agonists (Patwardhan et al., 2005; Berg et al., 2007a). This effect was dependent on integrins colocalized with opioid receptors in the neuronal membranes (Berg et al., 2007b). Likewise, painful paw inflammation and activation of sensory neurons by capsaicin or P2Y receptor agonists enhanced membrane recruitment of δ-receptors (Bao et al., 2003; Gendron et al., 2006). In vivo, local pretreatment with bradykinin or arachidonic acid enabled peripheral δ-opioid antinociception (Rowan et al., 2009). Another group showed that pretreatment of cultured DRG neurons with proinflammatory chemokines resulted in internalization and functional inhibition of μ-receptors (Zhang et al., 2004). Whether observations in neuronal cultures (i.e., cell bodies) reflect the scenario at peripheral terminals of DRG neurons needs to be investigated.
Peripheral tissue inflammation can induce differential regulation of opioid receptor types and their respective mRNAs in DRG neurons. μ-Receptors were most extensively studied and were consistently shown to be up-regulated (for review, see Busch-Dienstfertig and Stein, 2010). In complete Freund's adjuvant-induced hindpaw inflammation, for example, μ-receptor mRNA displays a biphasic up-regulation (at 2 and 96 h), whereas mRNA for δ-receptors remains unchanged, and κ-receptor mRNA shows a peak at 12 h (Puehler et al., 2004, 2006). In parallel, μ- and κ-receptor binding is up-regulated. This up-regulation is dependent on neuronal electrical activity (Puehler et al., 2004), on cytokine production in the inflamed tissue (Puehler et al., 2006), and may be mediated by cytokine-induced binding of transcription factors to opioid receptor gene promoters (for review, see Kraus, 2009).
Subsequently, the expression of opioid receptors on DRG membranes is enhanced (Zöllner et al., 2003; Shaqura et al., 2004), and the peripherally (but not centrally) directed axonal transport of opioid receptors is significantly augmented (Hassan et al., 1993; Ji et al., 1995; Mousa et al., 2001; Ballet et al., 2003; Puehler et al., 2004). This transport is stimulated by cytokines and nerve growth factor produced within the peripheral inflamed tissue (Donnerer et al., 1992; Jeanjean et al., 1995; Mousa et al., 2007a) and results in an increased density of opioid receptors on peripheral nerve terminals (Stein et al., 1990b). Not surprisingly, short-lasting (30 min) inflammatory stimuli (intraperitoneal acetic acid) did not significantly change the amount of immunostained opioid receptors on peripheral sensory nerve terminals (Labuz et al., 2007). Thus, the expression and axonal transport of opioid receptors varies according to receptor type and duration of inflammation.
In DRG neurons from rats with hindpaw inflammation, the up-regulation of opioid binding sites was due to an increase in both the number of neurons expressing receptors and the number of receptors per neuron, whereas the receptor affinity of opioid agonists remained unchanged (Zöllner et al., 2003). In addition, G protein coupling of opioid receptors was augmented (Zöllner et al., 2003; Shaqura et al., 2004). Extracellular recordings from peripheral sensory nerve fibers demonstrated opioid inhibition of spontaneous and stimulus-evoked action potentials in models of tissue injury (Stein, 1993; Andreev et al., 1994; Junger et al., 2002; Wenk et al., 2006). Inflammation is also accompanied by a sprouting of opioid receptor-bearing peripheral sensory nerve terminals (Mousa et al., 2001) and by a disrupted perineural barrier facilitating the access of opioid agonists to their receptors (Antonijevic et al., 1995; Rittner et al., 2009b). In addition, low extracellular pH increases opioid agonist efficacy, presumably by altering the interaction of opioid receptors with G proteins and adenylyl cyclase (Rasenick and Childers, 1989; Selley et al., 1993; Vetter et al., 2006). Under conditions of elevated cAMP/protein kinase A activity (as seen in inflammation), morphine can inhibit TRPV1 translocation to the plasma membrane, which may lead to reduced neuronal excitability (Vetter et al., 2008). All of these mechanisms can contribute to the increased antinociceptive efficacy of opioids in peripheral inflamed tissue. These findings help to explain why the proximal perineural application of opioids along intact (noninjured) nerves did not reliably produce analgesic effects in clinical studies (Picard et al., 1997).
c. Influence of nerve damage.
Mechanical nerve injury resulting in neuropathic pain is another condition influencing opioid receptor expression in sensory neurons. Most animal models have used unilateral nerve damage. The most frequent ones include 1) transection (axotomy) of peripheral nerves, sometimes involving resection of a portion (3–5 mm); 2) spinal nerve ligation (SNL), where one or two lumbar (L5 and/or L6) spinal nerves are tightly ligated; 3) partial sciatic (or saphenous) nerve ligation (PSL), in which the dorsal third to half of the nerve is tightly ligated; 4) chronic constriction injury (CCI) of the sciatic (or saphenous) nerve, in which loose ligatures (four in rats or two to three in mice) are placed around the nerve (for references see Machelska, 2011b).
Alterations in opioid receptor expression occur at both the mRNA and protein levels. After sciatic nerve transection (Zhang et al., 1998b) and SNL (Kohno et al., 2005), μ-receptor mRNA was down-regulated, and the number of μ-receptor-expressing neurons was decreased in the ipsilateral DRG. Similar effects were observed after PSL (Rashid et al., 2004; Pol et al., 2006), although μ-receptor immunoreactivity (analyzed by Western blot) was enhanced in another study (Walczak et al., 2005). After CCI, there was no strict correlation between μ-receptor mRNA and protein levels, in that mRNA was unchanged (Truong et al., 2003) or down-regulated (Obara et al., 2009, 2010), whereas the number of μ-receptor-expressing neurons was enhanced (Truong et al., 2003) or unaltered (Kolesnikov et al., 2007). In the same model, a reduction (by 37–39%) of the stimulation of G protein coupling to μ-receptors by morphine, but not by endomorphins, was reported (Obara et al., 2010). κ-Receptor mRNA was unchanged after PSL (Pol et al., 2006), lowered after CCI (Obara et al., 2009), and enhanced in animals with hypersensitivity after transection of the sacral nerve (Sung et al., 2000). δ-Receptor mRNA was not altered after PSL (Pol et al., 2006) or down-regulated after CCI (Obara et al., 2009). All these changes were observed 2 to 28 days after surgeries in lumbar DRG ipsilateral to the nerve injury compared with the contralateral DRG of injured animals (Zhang et al., 1998b; Truong et al., 2003; Pol et al., 2006) or to DRG of naive or sham-operated animals (Zhang et al., 1998b; Sung et al., 2000; Rashid et al., 2004; Kohno et al., 2005; Walczak et al., 2005; Pol et al., 2006; Obara et al., 2009, 2010). δ-Receptor protein was examined in a CCI-resembling model, in which a loose polyethylene cuff is placed around the sciatic nerve (Mosconi and Kruger model) (Kabli and Cahill, 2007). At 14 days after surgery, there were no changes in the number of DRG neurons, but there was an increase in immunohistochemical staining intensity of neuronal δ-receptors ipsi- and contralateral to the injury compared with sham-operated animals. In summary, mRNA levels of all three opioid receptors were usually unchanged or down-regulated in the DRG, regardless of the type of nerve injury. This was not always correlated with protein expression. μ-Receptors were most often studied. They were down-regulated after nerve transection and SNL, decreased or increased after PSL, and unchanged or increased after CCI. δ-Receptors were up-regulated in the Mosconi and Kruger model.
Because there is no doubt that nerve injury affects opioid receptor expression in DRG cell bodies, the net protein level might depend on injury-induced receptor relocation along the peripheral neuronal processes. All three opioid receptors were detected using immunohistochemistry in sensory fibers coexpressing CGRP at the CCI site of the sciatic nerve trunk (Labuz et al., 2009). Furthermore, an up-regulation of μ- and δ-receptors was detected by Western blot at the nerve injury site (Truong et al., 2003; Kabli and Cahill, 2007), and enhanced μ-receptor-immunoreactivity was found in hindpaw skin innervated by the damaged saphenous nerve after PSL or CCI (Walczak et al., 2005, 2006). All these alterations were demonstrated 1 to 14 days after nerve injury. Thus, regardless of changes in the DRG, opioid receptor protein expression was enhanced in lesioned nerves and in paw skin in various neuropathic pain models. How these alterations relate to antinocieptive effects will be discussed in section II.C.5.b.
B. Endogenous Opioid Ligands
The endogenous ligands of opioid receptors are derived from three independent genes that give rise to three precursors known as proopiomelanocortin (POMC), proenkephalin (PENK), and prodynorphin. Appropriate processing yields the major representative opioid peptides β-endorphin (END), Met-enkephalin, and dynorphin A, respectively. These peptides and their derivatives exhibit different affinity and selectivity for the μ- (END, Met-enkephalin), δ- (enkephalins, END), and κ- (dynorphin) receptors (Table 1). (for review, see Zöllner and Stein, 2007). Two additional endogenous opioid peptides have been isolated from bovine brain: endomorphin-1 and endomorphin-2. Both peptides are considered highly selective μ-receptor ligands, but their genetic origin and precursors remain unknown (for review, see Fichna et al., 2007).
1. Opioid Peptides in Peripheral Sensory Neurons.
Evidence for expression of enkephalins and dynorphins in DRG and peripheral sensory nerves began to accumulate in the 1980s. These peptides were shown to be transported toward central and peripheral nerve terminals and were mostly demonstrated in cutaneous and synovial neurons (for review, see Busch-Dienstfertig and Stein, 2010). Endomorphins were also detected (Martin-Schild et al., 1998; Pierce et al., 1998; Mousa et al., 2002; Scanlin et al., 2008). Relatively few studies addressed the consequences of tissue damage. One report found neither PENK nor prodynorphin mRNA in DRG of normal or polyarthritic rats (Calzà et al., 1998). Others detected mRNA for POMC, PENK, or prodynorphin in DRG of normal rats (Pohl et al., 1994; Braz et al., 2001; Obara et al., 2009). During hindpaw inflammation, PENK mRNA was transiently down-regulated, whereas mRNA of POMC and prodynorphin were not altered (Obara et al., 2009). After CCI, mRNA was unchanged for POMC, increased for prodynorphin, and decreased for PENK (Obara et al., 2009). The modulation of pain was addressed using a modified herpes simplex virus encoding PENK. Its application to the hindpaws of polyarthritic rats led to an increased number of DRG neurons expressing PENK mRNA and to elevated Met-enkephalin levels, resulting in improved locomotion and reduced thermal hyperalgesia (Braz et al., 2001). In a CCI model, this vector enhanced the expression of Met-enkephalin in trigeminal ganglia and sensory fibers and partially attenuated mechanical sensitivity. This antinociceptive effect was mediated by peripheral opioid receptors (Meunier et al., 2005). However, the underlying mechanisms (e.g., neuronal automodulation after peptide release, intracellular effects) were not examined in any of these studies.
2. Opioid Peptide Expression in Immune Cells.
The discovery that opioid receptors on sensory nerves are up-regulated during subcutaneous inflammation prompted the search for endogenous ligands within inflamed tissue. POMC-related peptides have been described in immune cells of many species, including humans (Stein et al., 1997; Smith, 2003; for review, see Rittner et al., 2008). At first, those peptides were assumed to be related to interferon-α, which was disproved later (for review, see Busch-Dienstfertig and Stein, 2010). Two groups simultaneously demonstrated the presence of full-length POMC mRNA encoding all three exons in immune cells (Lolait et al., 1986; Westly et al., 1986). Many other studies in leukocytes, however, described only truncated POMC mRNAs lacking the 5′ untranslated region from the first and the signal sequence from the second exon (for review, see Busch-Dienstfertig and Stein, 2010). The lack of the signal sequence omits the correct routing of the translation products and their processing to authentic POMC peptides in the regulated secretory pathway (Clark et al., 1990; Cool and Loh, 1994). Some studies found that the expression of full-length POMC mRNA was suppressed in mature unstimulated leukocytes, but could be induced under pathological conditions (Westly et al., 1986; Buzzetti et al., 1989; Ohta et al., 2000). Cabot et al. (1997) and Sitte et al. (2007) showed that POMC transcripts containing the signal sequence and subsequent END production are up-regulated in lymphocytes from rats with painful paw inflammation. Others found that the production of END in human peripheral leukocytes was induced by corticotropin-releasing factor (CRF) and suppressed by glucocorticoids (Smith et al., 1986). In addition, full-length POMC mRNA was detected in mature monocytes/macrophages and lymphocytes after stimulation with concanavalin A, CRF, interleukin (IL)-1 and -2, or phorbolester in vitro (Stephanou et al., 1991; Lyons and Blalock, 1997).
PENK mRNA, Met-enkephalin, and the appropriate enzymes for post-translational processing of PENK were also detected in human and rodent leukocytes (Vindrola et al., 1994; LaMendola et al., 1997). Deletion of the gene coding for PENK resulted in the complete absence of Met-enkephalin in both the brain and T cells, indicating that this peptide derives from the same precursor in the nervous and immune systems (Hook et al., 1999). In fetal thymocytes PENK mRNA was constitutively expressed but became undetectable at birth and thereafter (Linner et al., 1996). An earlier study showed that PENK mRNA expression was restricted to CD4-positive T lymphocytes and was absent in mature cytotoxic T cells (Linner et al., 1991). Others demonstrated that PENK mRNA expression can be induced by concanavalin A and phorbol myristate acetate in mature T lymphocytes (Rattner et al., 1991) and by IL-4 in peripheral blood mononuclear cells (Kamphuis et al., 1997), similar to neuroendocrine cells (Eiden, 1987; Joshi and Dave, 1992). In vivo murine neutrophils and monocytes/macrophages expressed PENK mRNA in zymosan- or thioglycollate-induced experimental peritoneal inflammation (Chadzinska et al., 2001). Leu-enkephalin, dynorphin and endomorphins were demonstrated in various types of immune cells (Cabot et al., 2001; Mousa et al., 2002; Chadzinska et al., 2005; Labuz et al., 2006).
In peripheral tissues subpopulations of opioid peptide-containing cells include granulocytes, monocytes/macrophages and lymphocytes (Przewłocki et al., 1992; Cabot et al., 1997; Mousa et al., 2001, 2007b; Rittner et al., 2001, 2007a; Labuz et al., 2006, 2010; Verma-Gandhu et al., 2006; Zöllner et al., 2008). Most notably, regulatory T cells (a population suppressing inflammatory responses) were shown to express the PENK gene (McHugh et al., 2002; Zelenika et al., 2002).
3. Migration of Opioid Peptide-Producing Cells to Inflamed Tissue.
The recruitment of leukocytes from the circulation into inflamed sites begins with cell rolling along the endothelial wall mediated predominantly by selectins. Subsequently, leukocytes are activated by chemokines released from endothelial and inflammatory cells and are presented on the endothelium (Fig. 1). This leads to up-regulation and increased avidity of integrins, which mediate the firm adhesion of leukocytes to endothelial cells via, for example, intercellular adhesion molecule-1 (ICAM-1). Finally, leukocytes transmigrate through the endothelium (Fig. 1) mediated by, for example, platelet-endothelial cell adhesion molecule-1 (Ley et al., 2007).
In inflamed rat paws it was shown that l-selectin, integrin β2, and the CXC chemokine receptor 2 (CXCR2) are coexpressed by opioid peptide-containing leukocytes (Mousa et al., 2000; Brack et al., 2004c; Machelska et al., 2004). Blocking selectins, ICAM-1, integrins α4 and β2, or chemokines CXCL1 and CXCL2/3 in vivo substantially decreased opioid-producing immune cells in inflamed tissue or injured nerves (Machelska et al., 1998, 2002, 2004; Brack et al., 2004c; Labuz et al., 2009). In addition, the recruitment of opioid-containing cells is dependent on neurokinin-1 receptors (which bind substance P) (Rittner et al., 2007b), on sensory and sympathetic neurons (Kager et al., 2010), and may be regulated by adhesion to neurons (Hua et al., 2006). Central mechanisms also play a role because intrathecally administered analgesic doses of morphine led to a decreased number of END-positive leukocytes in inflamed rat paws (Schmitt et al., 2003). This was confirmed in a clinical study using epidural analgesia in patients undergoing surgery (Heurich et al., 2007). Thus, an effective central inhibition of pain apparently leads to a decreased recruitment of opioid-producing cells to injured tissues. Likewise, it was shown that intrathecal administration of adenosine receptor-1 agonists inhibits neutrophil accumulation in inflamed skin of rats (Bong et al., 1996). This effect was mediated by adenosine-induced suppression of spinal glutamate receptor activation. This neutrophil trafficking was independent of primary afferent and sympathetic neurons but required intact peripheral nerve connection (Sorkin et al., 2003).
4. Processing and Release of Opioid Peptides from Immune Cells.
In the pituitary, POMC processing begins as the nascent polypeptide enters the endoplasmic reticulum directed by the signal peptide (references in Mousa et al., 2004). The POMC prohormone is directed to the regulated secretory pathway at the trans-Golgi network by binding to the sorting receptor carboxypeptidase E. The prohormone convertases PC1/3 and PC2 cleave POMC within the trans-Golgi network. Initially PC1 cleaves into adrenocorticotropic and β-lipotropic hormones. Inactive pro-PC2, bound to the chaperone-like binding protein 7B2, is transported from the endoplasmic reticulum to downstream compartments, where it matures to active PC2. Thereafter, PC2 converts β-lipotropic hormone into β-melanocyte-stimulating hormone and END (Mousa et al., 2004). All these components are also present in immune cells. We detected END and POMC alone and colocalized with PC1, PC2, carboxypeptidase E, and 7B2 in leukocytes in the blood and in inflamed paw tissue (Mousa et al., 2004).
The release of opioid peptides from immune cells can occur spontaneously (Stein et al., 1993; Sitte et al., 2007; Zöllner et al., 2008; Rittner et al., 2009a) or upon stimulation by various mediators. In the pituitary, the predominant factor is CRF, which also exerts this effect on leukocytes (Smith et al., 1986). Later studies showed that CRF and IL-1β can stimulate the secretion of opioid peptides from leukocytes in a receptor-specific and calcium-dependent manner (Schäfer et al., 1994; Cabot et al., 1997, 2001), as in the pituitary. Subsequently, several other releasing agents were identified. Opioid-producing immune cells coexpress adrenergic-, formyl peptide-, chemokine-, CRF-, and IL-1β-receptors (Mousa et al., 1996, 2003; Binder et al., 2004; Rittner et al., 2006a, 2009a). Noradrenaline stimulates END release from leukocytes in an adrenergic receptor-specific manner (Kavelaars et al., 1990; Binder et al., 2004; Mousa et al., 2004). The source of noradrenaline is sympathetic nerve fibers located in proximity to these cells (Fig. 1) (Binder et al., 2004). Activation of CXCR2 (by its ligand CXCL2/3) or the formyl peptide receptor (by mycobacteria) on granulocytes leads to release of END and Met-enkephalin. This is dependent on inositol triphosphate receptor-triggered release of Ca2+ from the endoplasmic reticulum, (partially) on phosphoinositol-3-kinase, and on p38 mitogen-activated protein kinase (Rittner et al., 2006a, 2007a, 2009a,b) (Fig. 1). The opioid peptides are packaged into vesicular structures and translocated to the membrane upon stimulation (Mousa et al., 2004; Rittner et al., 2007a; Zöllner et al., 2008). In granulocytes, these structures have been identified as primary (azurophil) granules (Rittner et al., 2007a). Thus, the characteristics of opioid peptide release from immune cells are consistent with the regulated secretory pathway.
C. Modulation of Pain and Inflammation via Peripheral Opioid Receptors
1. Peripherally Acting Exogenous Opioid Agonists.
Whereas early attempts to demonstrate peripheral opioid analgesia in noninjured tissue produced controversial results, subsequent studies in models of pathological pain were much more successful (for review, see Stein, 1993; Stein et al., 2003). In models of peripheral inflammation, the local injection of low, systemically inactive doses of μ-, δ-, and κ-agonists produced analgesia that was dose-dependent, stereospecific, and reversible by selective opioid antagonists (Stein et al., 1989; Stein, 1993). Antinociception was also shown in models of neuropathic (see section II.C.5.), visceral, incisional, thermal, bone, and cancer pain (Stein et al., 2009; for review, see Busch-Dienstfertig and Stein, 2010). Consistent with the in vitro studies discussed in section II.A.2, coadministration of μ- and δ-agonists may act synergistically (Schramm and Honda, 2010).
These findings stimulated the development of novel opioid ligands acting exclusively in the periphery without central side effects (Stein et al., 2009; for review, see Busch-Dienstfertig and Stein, 2010). A common approach was the design of hydrophilic compounds to reduce the capacity for crossing the blood-brain-barrier. Among the first generation of compounds were the μ-agonist loperamide (originally known as an antidiarrheal drug) and the κ-agonist asimadoline (Machelska et al., 1999). In a model of neuropathic pain, systemically (subcutaneously) applied loperamide reversed mechanical allodynia by activation of peripheral μ-receptors (Guan et al., 2008). Peripheral restriction was further pursued with arylacetamide morphinan-based [nalfurafine, [8-(3,3-diphenyl-propyl)-4-oxo-1-phenyl-1,3,8-triazaspiro[4.5]dec-3-yl]-acetic acid (DiPOA)] and with peptidic compounds [Tyr-Arg-Phe-Lys-NH2 (DALDA)] (for reviews, see Schütz et al., 2003; DeHaven-Hudkins and Dolle, 2004; Chu et al., 2007). Some of these were subsequently abandoned, mostly because the chemical addition of hydrophilic residues led to reduced affinity at opioid receptors or because higher doses were able to penetrate the blood-brain barrier.
How efficacious are such compounds compared with traditional opioid analgesics? In fact, several studies indicate that a large proportion (50–100%) of the analgesic effects produced by systemically administered conventional opioids can be mediated by peripheral opioid receptors (Craft et al., 1995; Binder et al., 2001; Reichert et al., 2001; Lewanowitsch and Irvine, 2002; Shannon and Lutz, 2002; Fürst et al., 2005; Labuz et al., 2007; Gaveriaux-Ruff et al., 2011). For example, in a rat model of urinary bladder inflammation, the peripherally restricted antagonist naloxone methiodide attenuated the antinociceptive effects of systemically applied μ- and κ-receptor agonists by 50% and fully abolished the effects of a δ-agonist (Craft et al., 1995). The conditional knockout of δ-opioid receptors in primary afferent neurons completely abrogated the antinociceptive effects of a systemically applied δ-agonist (Gaveriaux-Ruff et al., 2011). In acute pain induced by intraperitoneal acetic acid, naloxone methiodide reduced antinociceptive effects of systemically administered μ-, δ-, and κ-receptor agonists by 57, 80, and 47%, respectively. In mouse models without inflammation, increased hot plate latencies after systemic morphine application were inhibited by 50% after naloxone methiodide treatment (Lewanowitsch and Irvine, 2002). Others reported that naloxone methiodide had no effect on opioid analgesia (Ramabadran, 1982; Ramabadran et al., 1982), but at much lower doses than those examined in the study by Lewanowitsch and Irvine (2002). Human studies indicate that opioid agonists that do not readily cross the blood-brain barrier (e.g., morphine-6-glucuronide) are beneficial in patients with visceral and neuropathic pain (Eisenach et al., 2003; Wallace et al., 2006; Mangel et al., 2008) and can have the same analgesic efficacy as conventional opioids, with significantly fewer central nervous system side effects (Tegeder et al., 2003; Hanna et al., 2005; van Dorp et al., 2008). Thus, one may expect that peripherally restricted opioid agonists are active in both acute and chronic pain. Their efficacy should be at least half of that produced by conventional opioid analgesics, with the added benefit of reduced central adverse effects.
2. Extrinsic Stimulation of Opioid Peptide Release from Inflammatory Cells.
When applied to peripheral injured tissue, all the releasing agents mentioned in section II.B.4 can produce opioid-mediated antinociceptive effects in vivo. Depending on the agent, stage, and type of inflammation, these effects are mediated by different opioid peptides (Schäfer et al., 1994; Machelska et al., 2003; Mousa et al., 2003; Binder et al., 2004; Brack et al., 2004c; Labuz et al., 2006, 2009). Recent studies have suggested that α2-adrenergic agonists and serine proteases can also induce antinociception via enhanced production and release of endogenous opioids in models of inflammation (Ansah and Pertovaara, 2007; Martin et al., 2009). Various immunosuppressive treatments (e.g., cyclosporine A, depletion of granulocytes, blockade of chemokines or formyl peptide receptors, anti-selectin, anti-ICAM-1) reduce opioid-containing cells, opioid-mediated antinociception, and/or baseline nociceptive thresholds in inflamed tissue (Schäfer et al., 1994; Machelska et al., 1998, 2002; Brack et al., 2004c; Rittner et al., 2006a, 2009a; Labuz et al., 2009). Conversely, by transfer of allogenic lymphocytes (Hermanussen et al., 2004) or granulocytes (Rittner et al., 2006a), such impaired antinociception can be restored.
Controversies have revolved around the question of whether some of the agents (cytokines, chemokines, noradrenaline) may actually induce pain (hyperalgesia). In this regard, it is important to consider the presence versus absence of inflammation. In noninflamed tissue, certain cytokines (e.g., TNF-α, IL-6) were found to induce hyperalgesia (for review, see Cunha and Ferreira, 2003). In addition, several chemokines were described to induce pain (e.g., CCL22, CXCL12) or to decrease analgesic effects of other compounds (e.g., CCL5, CXCL12) (Oh et al., 2001; Szabo et al., 2002). Noradrenaline had no effect or increased pain behavior (references in Binder et al., 2004). The most obvious explanation for these findings is that noninflamed tissue does not contain opioid-producing immune cells. Hence, short of immune cells bearing their receptors, these agents now act on different targets (e.g., neurons or blood vessels). It is therefore not surprising that a given agent can produce different effects depending on the presence or absence of inflammatory cells. It is noteworthy that the selective recruitment of granulocytes does not elicit pain responses, suggesting that these cells are more important for the inhibition than for the generation of hyperalgesia (Rittner et al., 2006b, 2009b). Another intriguing finding in noninflamed tissue is that activation of keratinocytes by endothelin and cannabinoid agonists can lead to release of END, which then acts at opioid receptors on primary afferent neurons to inhibit nociception (Khodorova et al., 2003; Ibrahim et al., 2005).
3. Intrinsic Induction of Opioid Peptide Release from Inflammatory Cells.
Stress is a well known stimulus triggering endogenous analgesic mechanisms (Willer et al., 1981; Terman et al., 1984). In rats with unilateral hindpaw inflammation, stress induced by cold-water swim elicits potent antinociception in inflamed paws but not in the contralateral noninflamed paws (Stein et al., 1990a; Machelska et al., 2003). Although both peripheral and central opioid receptors contribute at early stages of the inflammatory response (several hours), endogenous analgesia is mediated exclusively by peripheral opioid receptors at later stages (several days) in this model (Stein et al., 1990a,b; Machelska et al., 2003). Thus, peripheral opioid mechanisms of pain control become more prevalent with the progression and severity of inflammation. The most prominent opioid peptide involved is END, but Met-enkephalin, dynorphin, and endomorphins also contribute to attenuation of nociception in this model (Stein et al., 1990a; Machelska et al., 2003; Labuz et al., 2006). Endogenous triggers of this stress-induced analgesia are locally produced CRF and sympathetic neuron-derived catecholamines (Schafer et al., 1996; Machelska et al., 2003; Binder et al., 2004).
Several lines of evidence indicate that such endogenous analgesic effects are dependent on immune cells: Stress-induced analgesia in animals with paw inflammation can be abolished by cyclosporine A, by whole-body irradiation, or by depletion of monocytes/macrophages (Stein et al., 1990b; Przewłocki et al., 1992; Brack et al., 2004a). l-Selectin, integrin β2, and CXCR2 are expressed by opioid-producing leukocytes (Mousa et al., 2000; Brack et al., 2004c; Machelska et al., 2004). Consistent block of selectins, ICAM-1, integrins, CXCL1, or CXCL2/3 in vivo substantially decreases the number of opioid-containing cells in inflamed tissue and abolishes endogenous peripheral opioid analgesia (Machelska et al., 1998, 2002, 2004; Brack et al., 2004c). Stress-induced analgesia is also decreased by blockade of neural cell adhesion molecule, presumably by preventing the adhesion of opioid peptide-producing cells to peripheral nerves in inflamed tissue (Hua et al., 2006). Thus, adhesion molecules have an impact on pain by modulating the extravasation of opioid producing immune cells and/or their adhesion to sensory neurons. In addition, central mechanisms apparently influence the migration of opioid-containing cells and endogenous analgesia in peripheral injured tissue (Schmitt et al., 2003; Heurich et al., 2007).
It is noteworthy that in models of inflammation (Sitte et al., 2007; Inglis et al., 2008; Zöllner et al., 2008; Rittner et al., 2009a) and bone cancer (Baamonde et al., 2006) and in humans undergoing knee surgery (Stein et al., 1993), the local injection of opioid antagonists or antibodies into injured tissue was shown to exacerbate pain. This strongly indicates that opioid peptides are continuously released and counteract hyperalgesia elicited by the many known proinflammatory agents present in inflammation (for review, see Rittner et al., 2008). Thus, even though hyperalgesia typically prevails in inflamed tissue, this would be much more severe if opioid peptides were not present and tonically released to blunt pain at the same time.
4. Tolerance to Opioid Analgesics.
Long-term opioid treatment can result in loss of opioid receptor function (desensitization, tolerance) (for review, see Zöllner and Stein, 2007). Previous concepts of opioid tolerance have been revised extensively in view of recent findings on the regulation of intracellular receptor trafficking (Waldhoer et al., 2004; Moore et al., 2007). Three mechanisms are mainly associated with desensitization of GPCR: 1) receptor phosphorylation, 2) receptor internalization and sequestration, and 3) receptor down-regulation (i.e., a reduced total number of receptors by degradation). After initial agonist binding, intracellular opioid receptor phosphorylation increases the affinity for arrestin molecules. Therafter, arrestin-receptor complexes sterically prevent coupling between receptor and G proteins and promote receptor internalization via clathrin-dependent pathways (Moore et al., 2007). Finally, the receptor is either degraded in lysosomes or it is dephosphorylated (i.e., resensitized) and recycled to the cell membrane. Extensive studies on these processes led to the current concept that receptor internalization and rapid recycling prevent the development of tolerance (for review, see Waldhoer et al., 2004; Tappe-Theodor et al., 2007; Koch and Höllt, 2008; Busch-Dienstfertig and Stein, 2010) (Fig. 1).
Experimental studies on tolerance are often performed in cell culture systems or in the absence of painful tissue injury, which precludes extrapolation to the clinical situation. However, recent studies showed that in the presence of painful paw inflammation, rats undergoing prolonged treatment with morphine do not develop signs of tolerance at peripheral μ-opioid receptors. In DRG neurons of these animals, internalization of μ-receptors was significantly increased, whereas G-protein coupling of μ-receptors and inhibition of cAMP accumulation were preserved. Moreover, opioid receptor internalization and signaling were reduced and tolerance was restored when endogenous opioid peptides in inflamed tissue were removed by antibodies or by depletion of opioid-producing granulocytes, monocytes, and lymphocytes with cyclophosphamide (Zöllner et al., 2008). Thus, the continuous availability of endogenous opioids in inflamed tissue apparently increases recycling and preserves signaling of μ-receptors in sensory neurons, thereby counteracting the development of tolerance to exogenous opioids. In vitro studies in transfected cells also showed receptor endocytosis induced by END and a correlation between the internalization/signaling potency of opioid ligands and μ-opioid receptor resensitization (Koch et al., 2005). Other studies of prolonged peripheral opioid receptor activation in the presence of different types of inflammatory stimuli also indicated a lack of tolerance development (Tokuyama et al., 1998; Börzsei et al., 2008; Mangel et al., 2008). These findings imply that the use of peripherally acting opioid agonists for the prolonged treatment of inflammatory pain is not necessarily accompanied by tolerance.
5. Neuropathic Pain
a. Immune response and pain after nerve damage.
When nerves are infected (e.g., by herpes virus) or mechanically damaged (e.g., by nerve crush, stretch, compression, or amputation), activation and recruitment of mast cells, neutrophils, macrophages, and/or lymphocytes occurs at the site of nerve injury and/or in the DRG (Watkins and Maier, 2002; Thacker et al., 2007; for review, see Austin and Moalem-Taylor, 2010). Pain-generating properties of immune cells have been subject of numerous review articles (Watkins and Maier, 2002; Moalem and Tracey, 2006; Scholz and Woolf, 2007; Thacker et al., 2007; Uçeyler et al., 2009; Austin and Moalem-Taylor, 2010; White and Miller, 2010; Machelska, 2011b). In some (but not all) studies, neuropathic pain was attenuated in animals with genetically delayed influx of macrophages, by stabilization of neuronal mast cells, or by depletion of circulating neutrophils or neuronal macrophages. The role of T lymphocytes was assessed in athymic nude rats and mice, in CD4 knockouts, and in recombination-activating gene-1 knock-outs as well as mice with severe combined immunodeficiency. These animals developed less mechanical or thermal hypersensitivity compared with wild-type animals after CCI, transection of spinal nerves, or spared nerve injury (Thacker et al., 2007; for review, see Austin and Moalem-Taylor, 2010; Machelska, 2011b).
Pronociceptive actions of immune cells were mostly attributed to proinflammatory cytokines, such as TNF-α, IL-1β, and IL-6. These cytokines occur both in leukocytes and in Schwann, satellite glial, and neuronal cells (Watkins and Maier, 2002; for review, see Austin and Moalem-Taylor, 2010), but the relative contribution of each cell type has not been clarified. TNF-α, IL-1β, and IL-6 mRNAs or proteins were found at the site of nerve injury or in DRG neurons. IL-1 receptor type 1 mRNA was detected in normal DRG neurons, whereas TNF-α receptors were present in the injured nerve trunk and the DRG after CCI. Electrophysiological and behavioral studies reported enhanced neuronal discharges and mechanical sensitivity after (low-dose) TNF-α application to injured DRG. Treatment with thalidomide (to inhibit TNF-α synthesis), etanercept (to prevent TNF-α binding to its cellular receptors), or TNF-α- and IL-1β-neutralizing antibodies attenuated hypersensitivity after CCI, PSL or SNL, similar to findings in IL-1- or IL-6-knockout mice. In contrast, IL-6 applied into the paw inhibited spinal nociceptive responses in SNL, suggesting bimodal actions of IL-6 in neuropathic pain (Thacker et al., 2007; Uçeyler et al., 2009; for review, see Austin and Moalem-Taylor, 2010; Machelska, 2011b).
In summary, there is no doubt that immune cells and their proinflammatory mediators contribute to neuropathic pain. However, attenuation of hypersensitivity by anti-inflammatory or immunosuppressive treatments was mostly moderate and was usually achieved only when such treatments commenced at the inception of, but not after the establishment of, neuropathy or when nerve injury was induced in animals already lacking immune cells or proinflammatory mediators. Furthermore, the elevation of nociceptive thresholds in T-cell-deficient animals was not always correlated with the abundance of these cells and might have been related to secondary alterations (e.g., in other cell types) (for review, see Austin and Moalem-Taylor, 2010; Machelska, 2011b). Thus, it is questionable whether general immunosuppression or blocking a single proinflammatory mediator will significantly alleviate clinical pain, particularly in patients with established neuropathy.
b. Neuronal opioid receptors and antinociception.
How do alterations in opioid receptor expression (see section II.A.5.c) relate to peripheral opioid antinociception in neuropathic pain? The PSL-induced down-regulation of μ-receptors in the DRG (Rashid et al., 2004) was associated with a lack of effect of μ-agonists injected into the ipsilateral paw and of the peripherally restricted agonist DiPOA applied systemically (Aley and Levine, 2002; Rashid et al., 2004; Whiteside et al., 2004). However, insufficient dosing or injection volume may have accounted for the absence of antinociception in these studies (Aley and Levine, 2002; Rashid et al., 2004; Whiteside et al., 2004). Suppression of opioid receptor expression by a silencer factor has also been suggested in PSL (Uchida et al., 2010). Although μ-receptors were down-regulated in DRG after SNL (Kohno et al., 2005), morphine injected into the ipsilateral paw or systemic loperamide still exerted antinociceptive effects (Pertovaara and Wei, 2001; Guan et al., 2008). In the CCI model, numerous studies have reported antinociceptive effects of immune cell-derived opioid peptides and of exogenous μ-, δ-, and κ-agonists applied via various routes (intravenous, intraperitoneal, at the nerve injury site, into the ipsilateral paw) (Kayser et al., 1995; Keïta et al., 1995; Catheline et al., 1998; Walker et al., 1999; Pertovaara and Wei, 2001; Martinez et al., 2002; Truong et al., 2003; Obara et al., 2004, 2007, 2009; Kolesnikov et al., 2007; Guan et al., 2008; Labuz et al., 2009, 2010). In the Mosconi and Kruger model, δ-receptor up-regulation in the lesioned nerve paralleled peripheral δ-agonist-induced antinociception (Kabli and Cahill, 2007; for review, see Machelska, 2011a). In summary, there is no clear correlation between opioid receptor mRNA levels in DRG and peripheral opioid antinociception in models of neuropathic pain. Rather, the presence and up-regulation of opioid receptors in injured nerve trunks or ipsilateral peripheral tissues innervated by damaged nerves seems to be decisive.
However, the underlying mechanisms are still unclear. Some authors proposed that processes characteristic for inflammatory pain (e.g., immune cells and mediators, perineurial barrier disruption; see section II.A.5.b) are relevant in neuropathic pain as well (Catheline et al., 1998; Walker et al., 1999; Martinez et al., 2002). Indeed, nerve injury is accompanied by inflammation (for review, see Watkins and Maier, 2002; Scholz and Woolf, 2007; Austin and Moalem-Taylor, 2010; see also section II.C.5.a). Although immune responses are associated with both CCI and PSL (see section II.C.5.a), peripheral opioid antinociception was commonly observed after CCI but not after PSL. It is noteworthy that immune cells accumulate directly at the nerve lesion (i.e., at the nerve trunk) but usually not in peripheral tissues (e.g., paws) innervated by the damaged neurons (Perkins and Tracey, 2000; Labuz et al., 2009). However, a large number of studies reported antinociception after opioid injection into paws. In addition, although the blood-nerve barrier was disrupted at the nerve injury site after SNL, this was not examined in paws (Abram et al., 2006). Undoubtedly, opioid receptors at the peripheral terminals of proximally injured nerves are functional but further studies on ligand accessibility, affinity, coupling, and signaling of these receptors are needed.
c. Opioid peptide-producing immune cells and antinociception.
The evidence on beneficial effects of opioid-producing leukocytes in inflammatory pain (see sections II.B.2–4 and II.C.2–3) is in contrast to the currently dominating opinion on immune cells as generators of neuropathic pain (see section II.C.5.a). Our group challenged this view (Labuz et al., 2009, 2010). Similar to earlier findings (see section II.C.5.a), we found many CD45+ leukocytes infiltrating the site of nerve injury at early (2–3 days) and later (14–15 days) stages of CCI neuropathy. Monocytes/macrophages dominated at both stages, followed by granulocytes. T lymphocytes arrived only during the later phase. Approximately 30 to 40% of CD45+ cells contained END, Met-enkephalin, or dynorphin and coexpressed CRF receptors. μ-, δ-, and κ-Opioid receptors were expressed in sensory fibers of the injured nerves. Application of CRF at the nerve injury site (2 and 14 days after CCI) fully blocked mechanical hypersensitivity. These antinociceptive effects were opioid-mediated and dependent on the immigration of opioid-containing leukocytes at the site of nerve damage (Labuz et al., 2009). Furthermore, END-containing T lymphocytes mediated opioid antinociception in advanced neuritis (14 days after CCI), as demonstrated by use of severe combined immunodeficiency mice and subsequent transfer of wild-type cells (Labuz et al., 2010).
Thus, in painful inflammatory neuropathy, immune cells apparently exert dual actions. The attenuation of neuropathic pain by anti-inflammatory and immunosuppressive strategies (leukocyte depletion, cytokine blockade) most likely resulted from the prevention of early detrimental effects of leukocytes, as these treatments had to commence at the initiation of injury. Simultaneously, however, the antinociceptive actions of leukocyte-derived opioids may have been hindered. It is noteworthy that opioid-containing immune cells can be effective not only at the initiation but also after the establishment of painful neuropathy.
6. Opioid Effects on Inflammation and Wound Healing
The discoveries and mechanisms described so far have spawned two new fields of research, the effects of opioids on inflammation and on wound healing. Apart from peripheral sensory neurons, opioid peptides and opioid receptors are expressed in immune cells (see II.4), fibroblasts, melanocytes, and keratinocytes (Tegeder and Geisslinger, 2004; Sharp, 2006; Chen et al., 2008; for review, see Bigliardi et al., 2009; Stein et al., 2009; Busch-Dienstfertig and Stein, 2010).
Peripherally acting opioids inhibit neurogenic inflammation by decreasing the release of substance P and CGRP from peripheral terminals of sensory neurons (Yaksh, 1988). Furthermore, opioids can reduce proinflammatory cytokines and adhesion molecules (Belkowski et al., 1995; Wilson et al., 1998; Walker et al., 2000; Philippe et al., 2003), edema and plasma extravasation (Jin et al., 1999; Khalil et al., 1999; Romero et al., 2005; Jiménez et al., 2006; Börzsei et al., 2008), and radiological and histological parameters (Binder et al., 2000, 2001; Walker, 2003). In human tissue sample, opioids have been shown to reduce proinflammatory cytokines, matrix metalloproteinase, and synovial leukocyte counts (Raap et al., 2000; Takeba et al., 2001; Straub et al., 2008). Intra-articular morphine injection decreased the number of inflammatory cells in synovial fluid of human patients with chronic arthritis (Stein et al., 1999) and diminished joint swelling, synovial fluid protein content, and serum inflammatory parameters in horses with experimental synovitis in vivo (Lindegaard et al., 2010a). On the other hand, some studies found nonopioid proinflammatory effects of asimadoline (Machelska et al., 1999) and morphine (Perrot et al., 1999). However, the weight of the evidence clearly supports anti-inflammatory activity.
In addition, opioids have mitogenic properties in the regeneration of mucosa (Cho et al., 2003), promote re-epithelialization and keratinocyte migration (Szabo et al., 2001; Bigliardi et al., 2002; Küchler et al., 2010), and up-regulate cytokeratin and transforming growth factor-β receptors, both important components in wound healing (Tegeder and Geisslinger, 2004; for review, see Bigliardi et al., 2009). In ischemic wounds, locally applied opioids hastened wound closure increased granulation tissue and collagen formation, increased epidermal and dermal organization, and enhanced angiogenesis (Poonawala et al., 2005; Gross et al., 2009). The vascular endothelial-derived growth factor receptor Flk1 and nitric oxide were also up-regulated by opioids in rat (Poonawala et al., 2005) and human keratinocytes (Wolf et al., 2009). However, depending on the chronicity of the wound and the stage of the healing process, some groups reported retarding effects in models of excisional wounds (Rook et al., 2008) and corneal abrasions (Sassani et al., 2003). Several comprehensive reviews summarized these findings and mostly concluded that topically applied opioids have the potential to improve wound healing and pain, and that μ-, δ-, and κ-opioids may have differential effects (Sassani et al., 2003; Tegeder and Geisslinger, 2004; Schmelz and Paus, 2007; Chen et al., 2008; Bigliardi et al., 2009; LeBon et al., 2009; Stein et al., 2009).
III. Nonopioid Immune-Derived Modulators of Pain
A. Antiinflammatory Cytokines
The most studied anti-inflammatory cytokines in relation to peripheral pain control are IL-4 and IL-10. Both are released by T lymphocytes, mast cells, macrophages, or granulocytes and decrease levels of TNF-α, IL-1β, and IL-6 (Cunha and Ferreira, 2003; for review, see Austin and Moalem-Taylor, 2010). In models of actue inflammatory pain, IL-4, IL-10, or IL-13 decreased nocieptive behaviors. Some authors suggested a peripheral site of cytokine actions, although this has not been assessed directly. In a neuropathic pain model, a viral vector encoding IL-4 led to the expression of IL-4 protein in DRG neurons and reversed mechanical and thermal hypersensitivity. These effects were associated with decreased levels of IL-1β, prostaglandin E2, and phosphorylated p38 mitogen-activated protein kinase in the spinal cord (Hao et al., 2006). Splenocytes stimulated to produce IL-4, IL-10, and IL-13 decreased mechanical and thermal hypersensitivity after CCI (Moalem et al., 2004). In addition, IL-10 injected at the initiation and site of nerve damage diminished the number of endoneurial TNF-α-expressing cells and partially attenuated thermal hypersensitivity after CCI (Wagner et al., 1998). Thus, in addition to opioid-containing leukocytes, anti-inflammatory cytokines can be beneficial in pathological pain.
B. Cannabinoids
The cannabinoid system consists of CB receptors and their endogenous ligands. Two receptors (CB1 and CB2) have been cloned so far. Among the endogenous ligands, N-arachidonoylethanolamine (anandamide) and 2-arachidonoyl-glycerol (2-AG) are the best studied. They are synthesized on demand from polyunsaturated fatty acid precursors by hydrolyzing enzymes in response to Ca2+ influx or mobilization from intracellular stores. Anandamide is primarily inactivated by the fatty acid amide hydrolase (FAAH) to arachidonic acid and ethanolamine. A cellular uptake mechanism has also been suggested but remains a matter of ongoing controversy. 2-AG is metabolized to arachidonic acid and glycerol mainly by monoacylglycerol lipase, but FAAH is also involved (for review, see Di Marzo, 2009). Both receptors and their endogenous ligands are distributed across the key pain-modulating pathways of the nervous system in the brain, spinal cord and peripheral sensory neurons (Agarwal et al., 2007; Anand et al., 2009; Di Marzo, 2009; Lever et al., 2009; Nyilas et al., 2009).
1. Cannabinoid Receptors on Peripheral Sensory Neurons.
Similar to opioid recptors, CB receptors belong to the family of seven-transmembrane GPCRs that couple to Gi/o proteins, leading to inhibition of adenylyl cyclase and cAMP production. In peripheral sensory neurons, activation of CB1 receptors was shown to inhibit voltage-gated N-type Ca2+ channels. Independent of CB1 receptors, cannabinoids were also reported to inhibit 5HT3 receptors and voltage-gated Na+ channels, resulting in a hyperpolarizing shift of the steady-state inactivation voltage and to dephosphorylate TRPV1 channels, leading to their desensitization. These mechanisms were suggested to account for peripheral antinociceptive effects of endocannabinoids and exogenous CB1 receptor agonists (for review, see Guindon and Beaulieu, 2009; Guindon and Hohmann, 2009; Kress and Kuner, 2009; Akopian et al., 2009; Sagar et al., 2009; McDougall, 2011). Direct evidence on peripherally mediated CB1-induced antinociception came from mice selectively lacking CB1 receptors in peripheral sensory (Nav1.8-expressing) neurons. These animals were more sensitive to noxious stimuli at baseline in models of visceral, inflammatory, and neuropathic pain and did not show antinociception upon CB1 agonist application (Agarwal et al., 2007). A peripherally restricted FAAH inhibitor elevated levels of anandamide in peripheral tissue, but not in the brain, and suppressed hypersensitivity in inflammatory and neuropathic pain in a CB1 receptor-dependent manner (Clapper et al., 2010). Peripheral CB2 receptors were also implied in alleviation of incisional, inflammatory and neuropathic pain (Kress and Kuner, 2009; for review, see Anand et al., 2009; Sagar et al., 2009; McDougall, 2011).
In addition to the above mechanisms, activation of CB1 receptors modulated ionotropic purinergic 2X and 2X2/3 receptors in nodose ganglia and DRG neurons in culture. The effects were usually (partially) inhibitory but in some neurons were facilitatory (for review, see Kress and Kuner, 2009). It is noteworthy that anandamide can also activate TRPV1 channels (for review, see Starowicz et al., 2007). This endocannabinoid triggered the release of CGRP from perivascular terminals of primary sensory neurons (Zygmunt et al., 1999) and the influx of Ca2+ in cultured DRG cells (Fischbach et al., 2007) in a TRPV1 antagonist-reversible manner. Other studies in cultured DRG neurons reported biphasic effects of anandamide. Thus, the release of CGRP or Ca2+ influx was decreased in a CB receptor-dependent manner by low concentrations of anandamide but was enhanced in a TRPV1-sensitive fashion by higher anandamide amounts (Tognetto et al., 2001; Ahluwalia et al., 2003). Anandamide also evoked excitation of C and Aδ nociceptors innervating healthy and arthritic rat joints in in vivo electrophysiological recordings (Gauldie et al., 2001). In addition, TRPV2-mediated CGRP release and transient receptor potential ankyrin 1-mediated intracellular Ca2+ elevation in response to plant-derived cannabinoids and/or endocannabinoids were shown in cultured DRG neurons (De Petrocellis et al., 2008; Qin et al., 2008). Thus, there is a potential for pronociceptive actions of cannabinoids, but the relevance of these effects to pain modulation in vivo needs to be clarified.
2. Cannabinoid Receptors on Immune Cells.
Although CB2 receptors seem to outnumber CB1 receptors in immune cells, both are expressed on splenocytes, lymphocytes, natural killer cells, mast cells, monocytes, macrophages, and neutrophils. Similar to neurons, CB receptor stimulation leads to down-regulation of cAMP formation. In vitro and ex vivo, both exo- and endocanabinnoids were shown to modulate cytokine and immunoglobulin production and/or release, antigen presentation, cell proliferation, migration, apoptosis, phagocytosis, and cytotoxic activity. In most cases, cannabinoid effects seemed immunosuppressive, but stimulatory actions were also described (for review, see Tanasescu and Constantinescu, 2010). In vivo, exogenous cannabinoids produced anti-inflammatory actions (for review, see Anand et al., 2009; Tanasescu and Constantinescu, 2010). A possible relevance of such effects to pain was suggested in a study using palmitoylethanolamide, a fatty acid amide with cannabinomimetic properties. Antinociception after application of palmitoylethanolamide, but not of anandamide, was associated with CB2 receptor-dependent attenuation of neutrophil migration to rat paws injected with nerve growth factor (Farquhar-Smith and Rice, 2003). The significance of such immunomodulatory effects in pain transmission has yet to be established.
3. Endogenous Cannabinoids in Immune Cells.
In view of peripheral antinociceptive actions of endocannabinoids (Guindon and Beaulieu, 2009; Guindon and Hohmann, 2009; Clapper et al., 2010), it would be interesting to identify their cellular source(s) in peripheral tissues. Similar to neurons, an endocannabinoid biosynthetic pathway has been described in immune cells (mostly macrophages). Macrophage cell lines or native cultured macrophages produced and secreted anandamide in response to Ca2+ ionophores, lipopolysaccharide, platelet-activating factor, exogenous Δ9-tetrahydrocannabinol, and anandamide itself. The effects of Δ9-tetrahydrocannabinol involved CB2 receptors (Bisogno et al., 1997; Pestonjamasp and Burstein, 1998). In addition to anandamide, monocyte/macrophage and mast cell/basophil cell lines produced and released palmitoylethanolamide. Both substances were taken up and degraded, although the inactivating enzyme was not identical to neuronal FAAH (Bisogno et al., 1997). 2-AG was also shown to be produced and secreted from various cell lines and native macrophages in response to lipopolysaccharide and/or platelet-activating factor, and it was inactivated by FAAH (Di Marzo et al., 1999). Macrophage-derived anandamide and 2-AG interacting with vascular CB receptors were implied in endotoxin-induced hypotension (Varga et al., 1998; Liu et al., 2003). However, the contribution of immune cell-derived endocannabinoids to pain modulation has not yet been examined.
IV. Therapeutic and Scientific Perspectives
A. Treatment of Pain and Inflammation
1. Topical and Intra-Articular Opioid Application.
Peripheral mechanisms of opioid analgesia have gained recognition in the clinical setting. Opioid receptors were demonstrated on peripheral terminals of sensory nerves in human synovia (Stein et al., 1996; Mousa et al., 2007b), dermal and epidermal nerve fibers (Ständer et al., 2002), and in dental pulp (Jaber et al., 2003). Analgesia after local (topical) opioid administration has been amply demonstrated in patients with various types of acute (e.g., orthopedic, dental, abdominal, urological, and eye surgery) and chronic pain (e.g., rheumatoid and osteoarthritis, oral mucositis, complex regional pain syndrome) (Azad et al., 2000; Dionne et al., 2001; Stein et al., 2003; Faktorovich and Basbaum, 2010). Several comprehensive reviews summarized these findings and mostly concluded that topically applied opioids have a potential to improve pain, albeit many clinical trials are small (Sawynok, 2003; Kopf et al., 2006; Bigliardi et al., 2009; LeBon et al., 2009).
The most studied and most successful application is the intra-articular injection of morphine into inflamed knee joints, which has been shown to produce analgesia with efficacy similar to that of local anesthetics or steroids without systemic or local side effects (Stein et al., 1991, 1999; Khoury et al., 1992; Likar et al., 1997, 1999, 2004). Veterinary trials of intra-articular morphine have demonstrated significant analgesic effects in dogs (Day et al., 1995; Sammarco et al., 1996) and horses (Santos et al., 2009). Further studies have demonstrated anti-inflammatory effects (Stein et al., 1999; Lindegaard et al., 2010b) and a lack of local tissue toxicity (Todhunter et al., 1996; Tulamo et al., 1996; Jaureguito et al., 2002). After intra-articular injection, morphine was detectable in synovial fluid for up to 24 h but did not reach clinically significant concentrations in the systemic circulation (Lawrence et al., 1992; Tulamo et al., 1996; Richardson et al., 1997; Brandsson et al., 2000; Lindegaard et al., 2010a).
The replication of such findings by many groups, meanwhile, has led to the establishment of peripheral opioid injections in clinical practice guidelines and curricula in human and veterinary medicine (American Society of Anesthesiologists Task Force on Acute Pain Management, 2004; Valverde and Gunkel, 2005). Several reviews have extensively analyzed the literature on intra-articular opioids. Although the majority agree on the analgesic efficacy of intra-articular morphine (Gupta et al., 2001; Kalso et al., 2002; Sawynok, 2003), some do not (Rosseland, 2005). Most commonly, negative results have been attributed to lack of study sensitivity (i.e., patients did not have sufficiently high baseline pain scores to detect a significant reduction), lack of tissue inflammation, or the superimposition of general or local anesthetic effects (Kalso et al., 2002; Rosseland, 2005; Stein, 2006). In addition, controversial conclusions from reviews can result from biased selection of studies (Stein, 2006).
2. Novel Peripherally Restricted Opioids.
Novel peripherally restricted κ-agonists were investigated in humans with chronic painful pancreatitis (Eisenach et al., 2003), neuropathic pain (Wallace et al., 2006), irritable bowel syndrome (Mangel et al., 2008), and after abdominal surgery (Menzaghi et al., 2010). These trials showed significant analgesia without central side effects. Morphine derivatives that do not cross the blood-brain barrier (morphine-6-glucuronide) were equieffective to morphine at pain relief but with significantly fewer side effects (e.g., sedation, respiratory depression) (Tegeder et al., 2003; Hanna et al., 2005; van Dorp et al., 2008). Although these compounds are promising, further research is needed into innovative ways of peripheral restriction while assuring analgesic efficacy.
3. Endogenous Opioids and Tolerance.
Opioid peptides were detected in human subcutaneous and synovial cells, mast cells, granulocytes, lymphocytes, and macrophages. The prevailing peptides are END and Met-enkephalin, but dynorphin and endomorphins were also found (Stein et al., 1993, 1996; Likar et al., 2004, 2007; Heurich et al., 2007; Mousa et al., 2007b; Rittner et al., 2007a; Straub et al., 2008). In patients undergoing knee surgery, blockade of intra-articular opioid receptors by the local administration of naloxone resulted in significantly increased postoperative pain (Stein et al., 1993). These findings suggest that in a stressful (e.g., postoperative) situation, opioids are tonically released in inflamed tissue and activate peripheral opioid receptors to attenuate clinical pain. In addition, CRF receptors are coexpressed with END in synovial inflammatory cells, and the intra-articular application of CRF can transiently reduce postoperative pain (Likar et al., 2007). Apparently, endogenous immune cell-derived opioids do not interfere with exogenous agonists, because intra-articular morphine is an equally potent analgesic in patients with and without opioid-producing inflammatory synovial cells (Stein et al., 1996; Likar et al., 2004). In line with our animal studies, this indicates that immune cell-derived opioids do not produce cross-tolerance to exogenous opioid agonists, but may, in fact, prevent the development of tolerance at peripheral opioid receptors (Zöllner et al., 2008). Additional evidence for this notion has been provided using a peripherally acting κ-agonist in patients with irritable bowel disease, where a significant reduction in pain was observable over a treatment period of 3 months (Mangel et al., 2008). This implies that the use of peripherally acting opioid agonists for the prolonged treatment of inflammatory pain is not necessarily accompanied by tolerance. This would be a major asset for long-term use in chronic inflammatory pain (for review, see Lang et al., 2010).
B. Future Directions and Knowledge Gaps
1. Clinical Perspectives.
A future challenge is to identify factors that selectively increase homing of opioid peptide-producing cells to injured tissue. For example, we showed that hematopoetic growth factors mobilized granulocytes in the blood, but this produced only a minor increase in the number of opioid-producing leukocytes in inflamed paws and no change of CRF- or cold water swim stress-induced antinociception (Brack et al., 2004b). Increasing the recruitment of opioid-producing cells with local injections of CXCL2/3 did not result in stronger antinociception either. This may have been a result of neutrophil-mediated oxidation of opioid peptides (Nagy et al., 2010) or of the relatively low number of neuronal opioid receptors at the respective (early) stage of tissue injury (Brack et al., 2004d). Indeed, our previous studies have shown that intrinsic analgesia increases with the duration of inflammation, in parallel with the number of opioid-producing leukocytes, the number of peripheral opioid receptors, and with the efficacy of opioid receptor-G protein coupling in sensory neurons (Mousa et al., 2001; Rittner et al., 2001; Zöllner et al., 2003).
Immunocompromised patients (e.g., in AIDS, cancer, diabetes, multiple sclerosis) frequently suffer from painful neuropathies (Dworkin et al., 2003). These can be associated with intra- and perineural inflammation, with reduced intraepidermal nerve fiber density, and/or with low CD4+ lymphocyte counts (Polydefkis et al., 2003). Although most animal studies concentrated on pain-generating properties of immune cells (see section II.C.5.a), antinociceptive actions of opioid peptide-containing leukocytes in experimental neuropathy were recently reported (see section II.C.5.c). It is noteworthy that in a clinical pilot study, higher numbers of macrophages in nerve biopsies correlated with the absence of phantom pain in patients after leg amputation (Stremmel et al., 2005). Thus, it may be interesting to investigate the opioid production/release and the migration of leukocytes in such patients. The important role of adhesion molecules and chemokines in the trafficking of opioid peptide-producing cells indicates that antiadhesion or antichemokine strategies for the treatment of inflammatory diseases carry a risk to exacerbate pain and to develop tolerance to opioid analgesics. Indeed, clinical trials have already shown that anticytokine and antiadhesion molecule treatments can lead to neurological disorders, including neuropathies (Simmons, 2005; for review, see Fromont et al., 2009).
It would be highly desirable to develop approaches that selectively augment opioid peptide production and/or increase peripheral opioid receptor numbers in damaged tissue. Various gene therapeutic avenues have been investigated (Mata et al., 2002; Pohl et al., 2003; Beutler et al., 2005; Kyrkanides et al., 2007; Machelska et al., 2009). Using nonviral, nonplasmid minimalistic, immunologically defined gene expression vectors, we found variable increases in leukocytic END production and a moderate attenuation of mechanical hypersensitivity (Machelska et al., 2009).
It has not been thoroughly examined whether combinations of opioids (e.g., endogenous opioid peptides and exogenous opioids) interact in a synergistic fashion in peripheral injured tissue, although initial studies have provided some evidence (Stein et al., 1996; Likar et al., 2004; Schramm and Honda, 2010). The effect of coapplied END or Met-enkephalin with exogenous opioids could be investigated in animals or patients with painful diseases showing minor leukocyte infiltration, e.g., in osteoarthritis. Similar to studies already performed in immunosuppressed animals (Labuz et al., 2010), a possible approach is to transfer opioid-producing cells into opioid tolerant animals with inflammatory pain to examine whether tolerance can be reversed. In this context, it would also be interesting to find out whether opioid peptides derived from primary afferent neurons exert self-inhibition.
A further promising strategy to augment the effects of endogenously released opioid peptides may be the inhibition of their degrading enzymes. This would avoid unphysiologically high concentrations of exogenous agonists at the receptor. Among peptidases localized on the surface of neurons and immune cells, the best characterized are neutral endopeptidase and aminopeptidase N, which inactivate opioid peptides (mainly enkephalins) and other substrates (Roques, 2000). Initial studies indicated that systemically (intravenously) administered dual inhibitors of both peptidases can produce antinociception involving peripheral opioids (Maldonado et al., 1994). It would be interesting to identify cellular sources of these enzymes and analyze levels of opioid peptides in inflamed tissue.
Relatively new fields are peripheral opioid effects on inflammation and wound healing. Although there is ample evidence from basic research (see section II.C.6), clinical studies are lacking. Obviously this has implications for all inflammatory conditions, including rheumatoid and osteoarthritis, inflammatory bowel disease (e.g., Crohn's disease, colitis ulcerosa, irritable bowel disease), inflammatory skin and mucosal diseases (e.g., herpes zoster, oral mucositis, sunburn, skin ulcers, neurodermitis, psoriasis, acne, burn, and incisional wounds), or pulmonary diseases (e.g., asthma, dyspnea) (see section II.C.6).
2. Knowledge Gaps.
Many questions in basic and applied research related to this field have not been tackled. A vast field is the endocytic trafficking and processing of opioid receptors in peripheral sensory neurons, particularly the influence of inflammation and the implications for tolerance development, efficacy, and potency of agonists. The studies discussed above have only scratched the surface. Likewise, oligomers or differential signaling of opioid receptors and the influence of inflammation and nerve injury have not been studied in peripheral sensory neurons so far, although computational modeling of signaling pathways has been attempted (Blätke et al., 2010). Despite convincing data on the attenuation of neuropathy-induced hypersensitivity in vivo, the expression, axonal transport and signaling of opioid receptors on peripheral terminals of nociceptors after nerve injury have not been thoroughly examined. Only a few studies investigated activation of opioid receptors in the trunk of injured nerves (Truong et al., 2003; Labuz et al., 2009, 2010). Finally, the role of opioid receptors on leukocytes in pain modulation needs to be explored.
It is still unclear how opioid-containing immune cells are directed to injured tissue, whether this migration is dependent on inflammation versus pain (or both), and which signals specifically stimulate opioid production in these cells during ongoing pain. Will there ever be vaccination against pain? Does treatment with nonsteroidal anti-inflammatory drugs interfere with the immigration of opioid-containing cells and subsequent antinociception? Is somatotopic placebo analgesia mediated by interactions between opioid-containing immune cells and peripheral opioid receptors (Benedetti et al., 1999)?
Technology-oriented research is needed to find novel ways of peripheral restriction of opioid compounds. For oral application, such compounds should be able to penetrate the gastrointestinal but not the blood-brain barrier; for topical application, they should sustain slow release of opioids (e.g., using nanocarriers) (Küchler et al., 2010). Manipulating the perineurium of peripheral sensory neurons is another interesting approach that might be pursued (Antonijevic et al., 1995; Rittner et al., 2009b). Selectively peripherally acting opioid analgesics would undoubtedly be preferred for their lack of central side effects (respiratory depression, nausea, dysphoria, addiction, cognitive impairment) and of adverse effects typical of nonsteroidal anti-inflammatory drugs (gastric erosions, ulcers, bleeding, diarrhea, renal toxicity, thromboembolic complications) (for review, see Stein and Kopf, 2009).
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft [Grants KFO 100, MA 2437/2-1, MA 2437/4-1, STE 477/9-1, GRK 1258]; the Bundesministerium für Bildung und Forschung [Grants 01GZ0311, MedSys 0101-31P5783, ImmunoPain 01 EC 1004 C]; the European Society of Anaesthesiology; and the International Anesthesia Research Society.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Stein and Machelska.
Footnotes
This article is available online at http://pharmrev.aspetjournals.org.
doi:10.1124/pr.110.003145.
↵1 Abbreviations:
- 2-AG
- 2-arachidonoyl-glycerol
- CB
- cannabinoid
- CCI
- chronic constriction injury
- CGRP
- calcitonin gene-related peptide
- CRF
- corticotropin-releasing factor
- DiPOA
- [8-(3,3-diphenyl-propyl)-4-oxo-1-phenyl-1,3,8-triazaspiro[4.5]dec-3-yl]-acetic acid
- DRG
- dorsal root ganglion
- END
- beta-endorphin
- FAAH
- fatty acid amide hydrolase
- GIRK
- G protein-coupled inwardly rectifying K+ channel
- GPCR
- G-protein-coupled receptor
- ICAM-1
- intercellular adhesion molecule-1
- IL
- interleukin
- PENK
- proenkephalin
- POMC
- proopiomelanocortin
- PSL
- partial sciatic nerve ligation
- SNL
- spinal nerve ligation
- TNF
- tumor necrosis factor
- TRPV
- transient receptor potential vanilloid.
- © 2011 by The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵





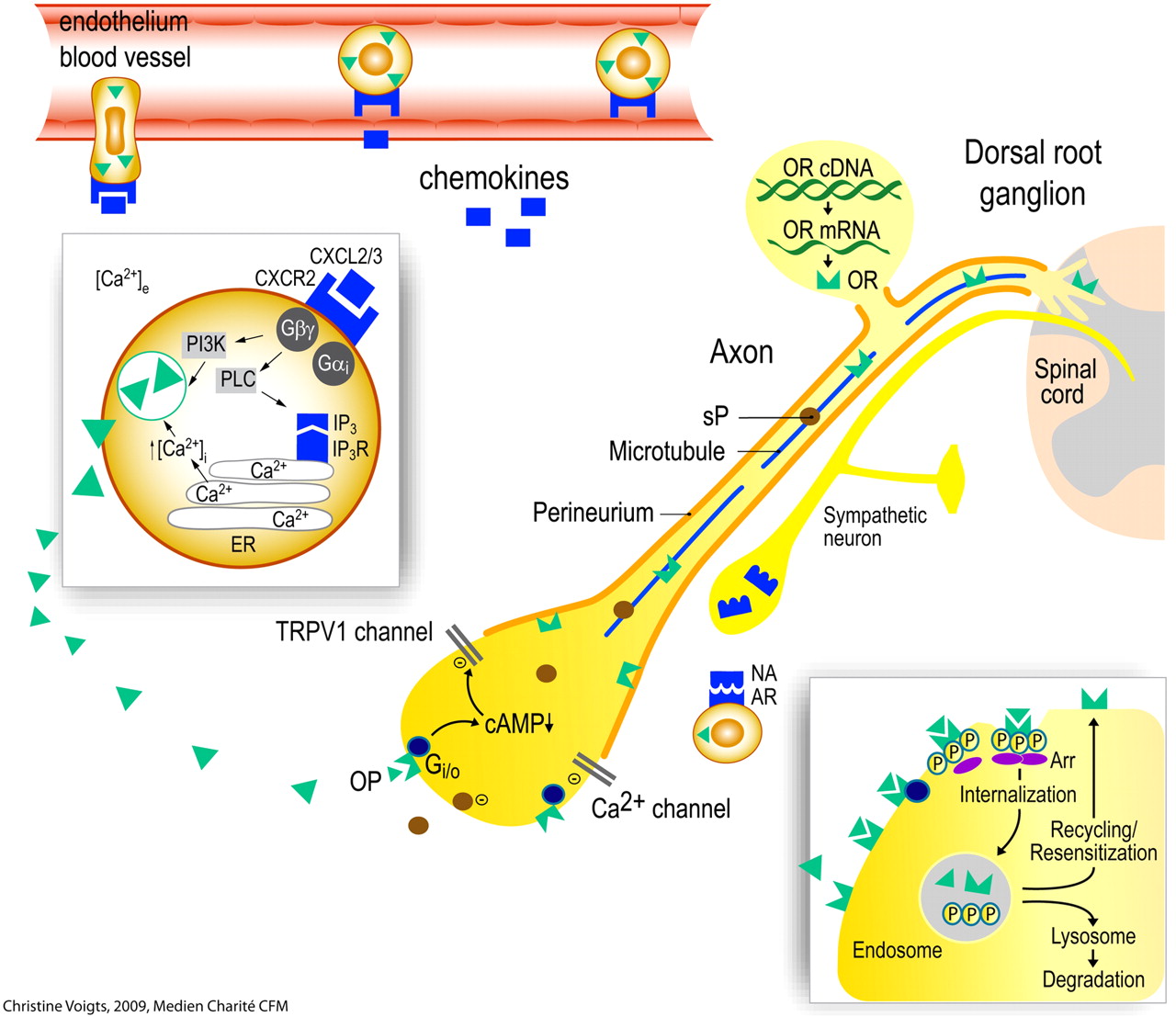{kind=link}