

Abstract
Objective. To evaluate the effects of etanercept treatment on patient-reported outcomes (PRO) in patients with psoriatic arthritis (PsA).
Methods. A 24-week double-blind comparison to placebo was followed by a 48-week open-label phase in which all eligible patients received etanercept. PRO were measured using the Stanford Health Assessment Questionnaire Disability Index (HAQ-DI), the Medical Outcomes Study Short-Form (SF-36), the EQ-5D visual analog scale (VAS), and the American College of Rheumatology (ACR) patient pain assessment.
Results. Beginning at Week 4 and continuing through Week 24 of double-blind treatment, patients treated with etanercept had significantly higher mean percentage improvement in HAQ-DI relative to baseline than patients given placebo (53.6% vs 6.4% at Week 24; p < 0.001). After 48 weeks of open-label treatment with etanercept, the mean percentage change from study baseline was 52.8% for the original etanercept group and 46.9% for the original placebo group, with 41.2% of patients overall achieving a HAQ-DI of 0. Mean changes relative to baseline for SF-36 physical component summary scores, EQ-5D VAS, and ACR pain assessment were also significant in the double-blind period for etanercept compared with placebo (p < 0.001 for all 3 measures). Patients taking placebo achieved similar improvements once they began treatment with etanercept in the open-label period.
Conclusion. Patients with PsA treated with etanercept reported significant improvements in physical function that were almost 10 times the improvement seen with placebo and were maintained for up to 2 years. Almost half of patients treated with etanercept reported no disability by the end of the study.
Standard evaluations of drugs used for the treatment of rheumatic diseases have been predominantly based on physician-evaluated outcomes. Although these measurements are critical to the determination of efficacy and safety of new drugs or new drug combinations, patient-reported outcomes (PRO), including assessments of physical disability, pain, global health, and health-related quality of life1–3, have emerged as important outcome measures in randomized clinical trials of patients with rheumatic diseases4–7. Such assessments quantify the patient’s judgment of the effect of treatment on their disease, thereby complementing and expanding physician-evaluated measures of disease severity. Use of both types of outcome measures allows a more complete evaluation of treatment response and aids in the interpretation of results, and ultimately improves decisions regarding future therapy2,3,6. Studies have suggested that PRO are as informative as, and more sensitive than, standard physician-reported measures8–12, and data from a metaanalysis of 3 rheumatoid arthritis (RA) trials suggest that PRO provide better discrimination of the treatment effect and are less likely to exhibit a placebo effect than traditional physician-reported outcomes5,13.
Although PRO have been studied in numerous RA trials4–7,14–16, less information regarding these endpoints is available for patients with psoriatic arthritis (PsA). One of the inflammatory arthritides, PsA is a chronic inflammatory condition estimated to occur in approximately 0.28% of the general population, with an estimated prevalence of 6%–39% in patients with psoriasis17–20. Although PsA shares some characteristics with RA, it frequently shows distinctive clinical features such as asymmetric involvement limited to a few joints, distal interphalangeal (DIP) joint involvement, axial involvement, enthesitis, dactylitis, and radiographic features such as pencil-in-cup deformity, gross osteolysis, joint space widening, ankylosis, juxtaarticular periostitis, shaft periostitis, and tuft resorption21–23. Many patients have erosive disease, physical limitations, and work-related disability22,24. Patients with PsA may experience more pain and more role limitations than patients with RA25,26 and patients with psoriasis alone27,28.
Although PRO are an important tool for physicians in the management of PsA, currently there is not one comprehensive instrument in use that adequately captures all aspects of the disease. For example, the Health Assessment Questionnaire has been shown to be effective for measuring functional disability in PsA but not for determining the psychosocial impact of the disease29,30. Therefore, the use of multiple PRO to identify disease-related sequelae is common and will likely remain so until PsA disease-specific instruments have been validated and used in clinical trials.
Etanercept, a high-affinity soluble tumor necrosis factor receptor blocker, has demonstrated effectiveness in improving clinical and radiographic outcomes, as well as PRO, in patients with RA14,15,31–35. Etanercept has also been shown to improve clinical and radiographic outcomes in patients with PsA in a randomized, placebo-controlled, multicenter trial and subsequent open-label followup24,36. In this trial, etanercept was also evaluated for its ability to improve physical function and pain in patients with PsA. Published results from the primary study indicate that patients treated with etanercept had significant improvement in the Stanford Health Assessment Questionnaire-Disability Index (HAQ-DI) and the Medical Outcomes Study Short-Form (SF-36) physical component score compared with placebo after 24 weeks of double-blind treatment36. Our objectives are to describe in detail the final results for patient-reported assessments across all measured PRO and times in the primary study.
MATERIALS AND METHODS
Study design
Details of the study design have been described (NCT00317499)36. There were 3 study phases, a 24-week double-blind phase, a 24-week blinded maintenance phase, and a 48-week open-label extension. Patients were randomly assigned to receive etanercept 25 mg subcutaneously twice weekly or placebo in the 24-week double-blind phase. Patients receiving methotrexate who were going to continue methotrexate were randomized separately from those who were not receiving methotrexate. During the maintenance phase, patients continued to receive blinded treatment for up to 24 weeks until all patients had completed the 24-week blinded phase and the database was locked. Patients were then eligible to participate in a 48-week open-label extension during which all patients received etanercept. The protocol was approved by the institutional review board of each participating center.
Patients
Eligible patients were 18 to 70 years of age and diagnosed with PsA with active arthritis and an inadequate response to nonsteroidal antiinflammatory drug (NSAID) therapy at the time of entry. The study excluded patients who were pregnant or breastfeeding, as well as those with diabetes mellitus requiring insulin, uncompensated congestive heart failure, angina pectoris, uncontrolled hypertension, severe pulmonary disease requiring therapy, and history of cancer other than resected cutaneous basal and squamous cell carcinoma or in situ cervical cancer. All patients provided written informed consent before study entry. Disease-modifying antirheumatic drugs (DMARD) were not permitted during the study. Methotrexate, NSAID, corticosteroids, and topical therapies were permitted if stable doses had been achieved before study entry.
Study drug
The study medication (etanercept or placebo) was supplied as sterile, lyophilized powder in vials and was reconstituted by site personnel not involved in data collection for the study (double-blind phase) or by patients (open-label phase). Study medication was administered twice weekly by subcutaneous injection either by the patient or a designated person. Change in dose of study drug was not permitted. Interruptions of up to 1 week (i.e., 2 doses) were permitted in the case of grade 3 or 4 toxicity, defined according to the National Institutes of Health, National Cancer Institute Common Toxicity Criteria (version 2.0). Recurrence of the same grade 3 or 4 toxicity necessitated discontinuation of treatment.
Clinical and radiographic assessments
Efficacy of treatment of arthritis was measured by the percentages of patients meeting the American College of Rheumatology 20% response criteria (ACR20), as modified for PsA by inclusion of DIP and carpometacarpal (CMC) joints, and the Psoriatic Arthritis Response Criteria (PsARC)36,37. Response to treatment of psoriasis was based on the dermatologist’s global assessment of target lesions and the percentage of patients achieving 50% improvement on the Psoriasis Area and Severity Index (PASI 50)36. Articular damage was assessed using total Sharp scores of radiographic images of the hands and wrists; Sharp scores were modified for PsA by inclusion of the DIP and CMC joints.
Patient-reported outcomes
Patient-reported outcomes included the HAQ-DI38,39, the SF-3640,41, the EQ-5D visual analog scale (VAS) (i.e., feeling thermometer1) and the ACR patient pain assessment42. The HAQ-DI comprises a series of 20 questions in 8 domains (dressing and grooming, arising, eating, walking, hygiene, reach, grip, and activities) related to physical functioning as required for usual daily activities. The HAQ-DI score is a composite ranging from 0 to 3, with lower scores indicating better outcomes.
SF-36 scores are based on a 36-item questionnaire measuring 4 physical function domains (physical functioning, role limitations attributable to physical problems, bodily pain, and general health) and 4 mental function domains (vitality, social functioning, role limitations attributable to emotional problems, and mental health). Scores for each domain ranged from 0 to 100, with higher scores indicating better functional status. SF-36 scores were normalized according to the method of Ware and colleagues43, which uses data from the general US population, in which 50 points represents the US norm and 10 points represents 1 SD unit.
The EQ-5D VAS evaluated the patient’s health state using a vertical VAS ranging from 0 (worst health) to 100 (best health). The ACR patient pain assessment used a horizontal Likert scale, with 0 corresponding to “normal” and 10 corresponding to “most abnormal.” All PRO were measured at baseline and at 4, 12, and 24 weeks during the double-blind period; every 12 weeks during the maintenance period; and every 12 weeks during the 48-week open-label period. To gauge patient assessment of change, patients were asked to rate the importance of their change in physical function on a 7-point scale (1 = not at all important; 7 = extremely important).
Statistical analysis
Treatment group comparisons were based on scores and percentage change for the HAQ-DI and ACR pain scale, as well as normalized scores and change from baseline for the EQ-5D VAS and SF-36 physical component and mental component summary scores. The percentage improvement from baseline was defined as the percentage change in the direction of improvement, that is, lower HAQ-DI and ACR pain scores and higher SF-36 and EQ-5D VAS scores. Hypothesis testing of treatment comparisons was based on a 2-sided Wilcoxon rank-sum test. No adjustments were made for multiple comparisons. In the double-blind treatment period, missing data were imputed using the method of last observation carried forward. In the open-label phase, analyses were based on observed patients; no imputation was performed.
RESULTS
Patients
A total of 205 patients were randomized in the double-blind phase of the study: 101 were randomly assigned to etanercept and 104 were randomly assigned to placebo. Of these, 169 (88 originally assigned to etanercept; 81 originally assigned to placebo) elected to participate in the open-label study. The 2 randomized groups were well balanced at baseline in terms of demographic and disease characteristics36. The median time since diagnosis of PsA was 6 to 7 years, the median age was approximately 47 years, and approximately 90% of patients were white. The placebo group had a slight predominance of women (55%), while the etanercept group had a slight predominance of men (57%). Patients in the etanercept group had more severe radiographic disease at baseline than patients in the placebo group. Most patients (84%) had polyarticular arthritis. The 169 patients who elected to participate in the open-label study had baseline demographic characteristics and disease history similar to those in the overall cohort of 205 randomized patients, as did the 148 patients who completed the 48-week open-label period23. Mean (range) total exposure to etanercept was 560.1 (71–744) days for patients originally randomized to etanercept and 315.5 (1–361) days for patients originally randomized to placebo.
Clinical efficacy
Detailed analyses of clinical and radiographic results and safety measurements in this study were reported elsewhere36. During the double-blind phase of the study, significantly more patients randomly assigned to etanercept responded, as determined by ACR20 criteria (p < 0.0001), PsARC (p < 0.001), and PASI 50 criteria (p < 0.001) than did patients randomly assigned to placebo. Radiographic disease progression was inhibited in the etanercept group compared with the placebo group at 12 months (p = 0.0001). Within 12 weeks of starting etanercept in the open-label phase of the study, patients originally assigned to placebo achieved comparable clinical results to patients initially randomized to etanercept treatment, and results were maintained in both groups throughout the open-label phase23.
Patient-reported outcomes
Mean HAQ-DI scores before treatment were 1.1 units in both groups. Within 4 weeks of initiating treatment, HAQ-DI scores were significantly improved in patients receiving etanercept compared with scores in patients receiving placebo. Week 4 scores were 0.7 units for etanercept and 1.0 unit for placebo, corresponding to percentage improvements of 35.1% and 8.0%, respectively (p < 0.001; Figure 1A). Continued improvement in HAQ-DI scores was observed with additional treatment (Table 1), reaching mean percentage changes of 53.5% in the etanercept group by Week 12, compared with a 6.3% change for patients in the placebo group (p < 0.001; Figure 1A). Improvements achieved at Week 12 were maintained at Week 24 in both groups (Table 1 and Figure 1A). At the start of the open-label phase, mean HAQ-DI scores were 0.4 units in the etanercept group and 1.0 unit in the placebo group. During the open-label phase, patients originally assigned to etanercept therapy maintained or improved their HAQ-DI scores from the double-blind period (Figure 1B) with a 52.8% improvement from original study baseline at Week 48. Patients originally assigned to placebo demonstrated improvement in their HAQ-DI scores within 12 weeks of initiating treatment with etanercept, reaching a mean score of 0.7 at Week 12 (36.2% improvement from original study baseline). Continued improvement was observed, with patients achieving a mean score of 0.6, corresponding to a 46.9% improvement at Week 48 (Table 1 and Figure 1B).
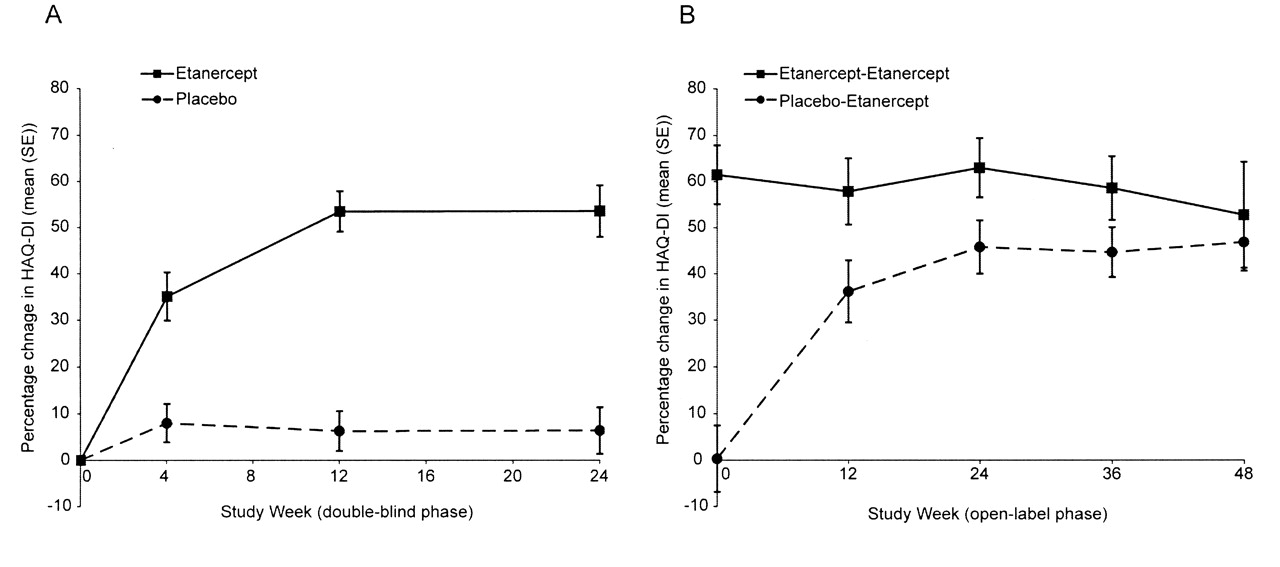
Mean percentage improvement in HAQ-DI scores. All data represent the mean percentage change from baseline and error bars are standard error. A. Double-blind phase: Results are based on last observation carried forward imputation; mean baseline scores were 1.1 (0.1) units for both treatment groups. B. Open-label phase: Results are based on the observed population at each time; baseline scores were 1.0 (0.1) units for patients taking placebo-etanercept and 0.4 (0.1) units for patients taking etanercept-etanercept. HAQ-DI: Health Assessment Questionnaire-Disability Index.
Mean scores for patient-reported outcomes during the double-blind and open-label treatment periods. Data given under “placebo” refer to placebo-etanercept during the open-label phase. Data under “etanercept” refer to continued etanercept during the open-label phase. No imputation was used for missing data during the open-label phase.
At the end of the double-blind phase, the percentage of patients with a HAQ-DI score of 0, indicating no disability on any of the 8 domains of the HAQ-DI, was higher among patients randomized to etanercept than among patients randomized to placebo (38% vs 7%; Figure 2). After 48 weeks of open-label treatment, 53% of patients continuously treated with etanercept and 29% of patients originally assigned to placebo reported a HAQ-DI of 0.
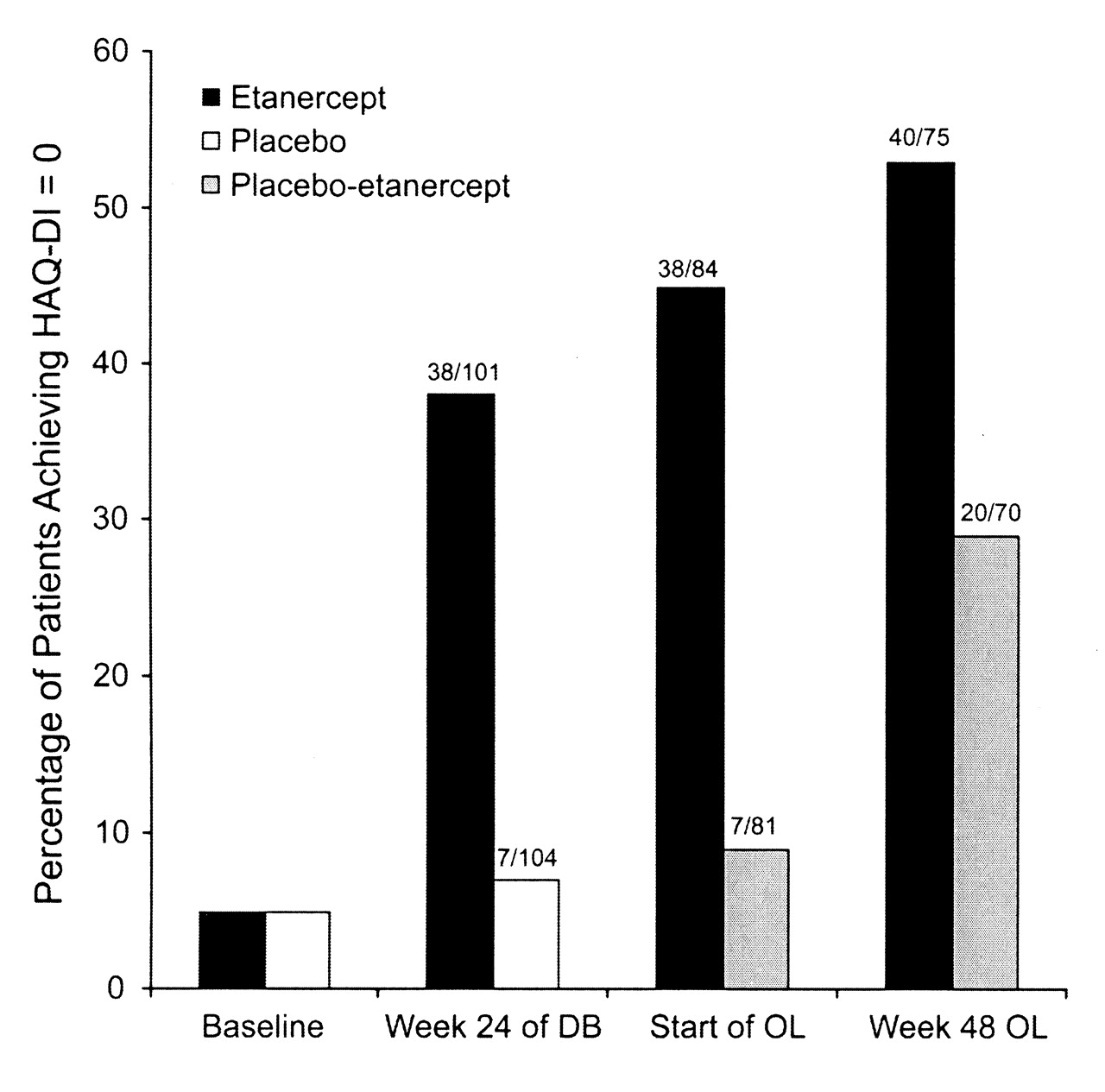
Percentage of patients with no disability according to HAQ-DI, defined as a HAQ-DI score of 0. Results are based on last observation carried forward imputation in the double-blind phase and on the observed population at each time in the open-label phase. Baseline refers to data obtained at study entry. HAQ-DI: Health Assessment Questionnaire-Disability Index; DB: double-blind phase; OL: open-label phase.
Before treatment, mean SF-36 physical component summary scores for patients in both treatment arms were below the US norm of 50 units (etanercept, 35.8 units; placebo, 35.7 units). During the double-blind phase, the mean improvement from baseline in SF-36 physical component summary scores was significantly greater for patients in the etanercept group than for patients in the placebo group as early as Week 4 (5.8 units vs 0.5 units; p < 0.001) and at all later assessment times (Figure 3A). By Week 24 of the double-blind phase, SF-36 scores had improved by 9.3 units in the etanercept group vs 0.7 units in the placebo group (p < 0.001; Figure 3A) to scores of 45.1 and 36.4, respectively (Table 1). SF-36 physical component summary scores at open-label baseline were 46.4 for patients originally assigned to etanercept and 36.8 for patients originally assigned to placebo. During the open-label phase, patients originally assigned to etanercept therapy maintained or improved their SF-36 physical component summary scores from the double-blind period (Table 1 and Figure 3B). Patients originally assigned to placebo improved their physical component summary scores by 8.3 units within 12 weeks of starting treatment with etanercept, and by the end of the 48-week open-label study period, these patients achieved a mean score of 44.1 units (Table 1).
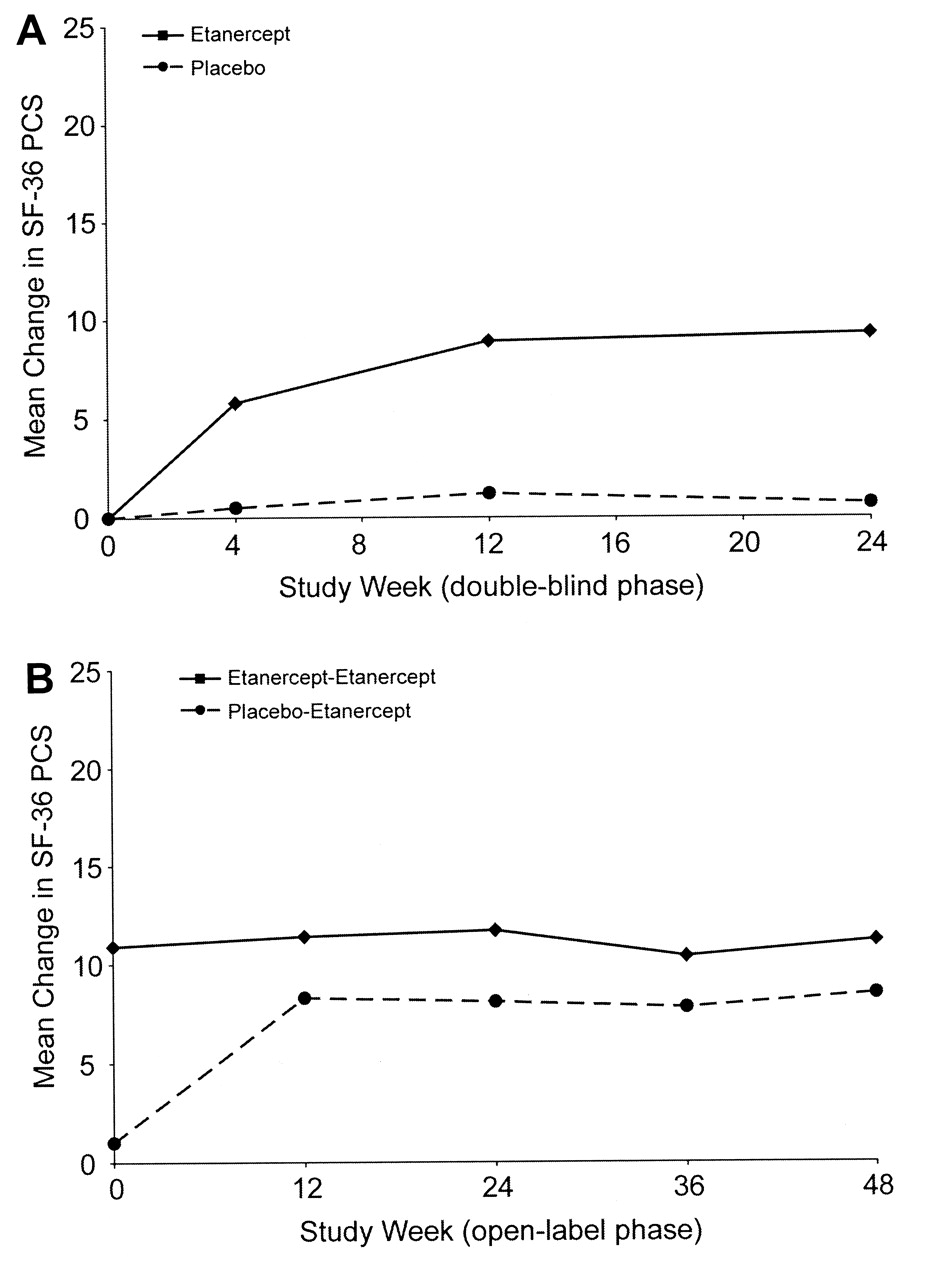
Mean change in SF-36 physical component summary scores. All data represent the mean change from baseline. A. Double-blind phase: SF-36 physical component summary scores were assessed at 4, 12, and 24 weeks; results are based on last observation carried forward imputation; mean baseline scores were 35.7 units for patients given placebo and 35.8 units for patients treated with etanercept. B. Open-label phase: SF-36 physical component summary scores were assessed at 12, 24, 36, and 48 weeks of treatment; results are based on the observed population at each time; baseline scores were 36.8 units for patients given placebo-etanercept and 46.4 units for patients taking etanercept-etanercept. SF-36 PCS: Short-Form 36 physical component summary.
Mean SF-36 mental component summary scores at original study baseline were at or approaching the US norm of 50 units in both treatment groups (etanercept, 50.9 units; placebo, 48.4 units; Table 1). As expected, mean changes in SF-36 mental component summary scores were of lower magnitude than the changes observed for SF-36 physical component summary scores in both groups of patients and in both treatment periods (Table 1). After 24 weeks of blinded therapy, changes of 2.7 units and −0.1 units (p = 0.062) were observed in the etanercept and placebo groups, respectively. Mean scores at the start of the open-label phase were 53.7 units for patients originally assigned to etanercept and 49.1 units for patients originally assigned to placebo (Table 1). Improvements from the original study baseline of 1.6 units and 0.6 units, in the etanercept-etanercept and placebo-etanercept groups, respectively, occurred after 48 weeks of open-label treatment.
Patients randomized to etanercept reported better global health according to the EQ-5D VAS after 24 weeks of blinded therapy than did patients randomized to placebo (Table 1). The mean change from baseline to 24 weeks was 14.3 units in patients taking etanercept and 2.1 units in patients taking placebo (p < 0.001). Patients taking etanercept maintained the improvement in EQ-5D VAS scores throughout the open-label period (Table 1). EQ-5D VAS scores of patients originally assigned to placebo reached parity with the etanercept group by 12 weeks of open-label treatment with etanercept and were maintained throughout 48 weeks.
A significant improvement in pain, as measured by the ACR patient pain assessment, was seen as early as 4 weeks in the etanercept group and continued to improve over 24 weeks of blinded therapy (Table 1). At 24 weeks, ACR pain scores had improved by a mean of 48.4% in the etanercept group, compared with a mean change of −1.1% in patients given placebo (p < 0.001) to scores of 1.6 and 2.8, respectively. Improvements in ACR pain scores were maintained in the etanercept group during the open-label phase (Table 1). ACR pain scores improved in the placebo group upon initiation of open-label etanercept therapy, with a mean score of 1.5, corresponding to 41.1% improvement, achieved at 48 weeks.
DISCUSSION
Patients with PsA experience significant functional impairment and considerable pain44. In our study, the longterm use of etanercept in patients with PsA resulted in clinically meaningful improvement in PRO, which included the HAQ-DI, SF-36 physical component score, EQ-5D VAS, and the ACR patient pain assessment scale. Patients receiving etanercept in the double-blind portion of the study reported rapid improvement in outcomes that were evident by the first assessment. Patients randomized to placebo who switched to etanercept treatment during the open-label phase achieved similar responses within 12 weeks, and improvements were maintained in both groups for up to 2 years of followup treatment.
The functional improvement in patients with PsA following etanercept treatment observed in our study is consistent with improvement observed in a Phase 2 study of etanercept treatment in patients with PsA37. In our study, patients receiving etanercept experienced rapid and sustained improvements in HAQ-DI and SF-36 physical component summary scores that were maintained throughout the treatment period. Mean SF-36 physical component summary scores were well below the US norm of 50 units at baseline and were improved to levels that approached the US norm with etanercept therapy. At baseline our patient population had a mean SF-36 mental component summary score that was at normal levels for the US population. Thus, as expected, SF-36 mental component summary scores did not significantly improve with etanercept treatment over the course of our study.
In addition to improving functional ability in patients with PsA, etanercept has been shown to reduce patient-reported pain. In a randomized clinical trial of etanercept in patients with PsA and psoriasis, patient-reported pain was eliminated in 17% of patients treated with etanercept, while patients who received placebo reported no pain cessation37. In our study, patients treated with etanercept in the double-blind period experienced substantial improvement in self-reported pain compared with patients receiving placebo. When patients in the placebo group were treated with etanercept in the open-label period, improvement in patient-reported pain was evident by the first assessment and continued throughout the duration of our study. The improvement in physical function and reduction in pain may have contributed to the steady and consistent improvement in patient-perceived health state, as measured by EQ-5D, in patients treated with etanercept over the course of our study.
Patient-reported outcomes are increasingly important in assisting physicians in making appropriate treatment decisions for patients with PsA. Although the instruments used in this analysis were not specifically developed for patients with PsA, they have been shown to be responsive, discriminative, and relatively easy to use29. Moreover, while no single instrument can accurately quantify the total effect of disease at present, the use of multiple patient self-reported instruments may help to determine the functional and psychosocial ramifications of PsA. In our study, we have shown that the extended treatment with etanercept in patients with PsA is not only effective with respect to traditional physician-based assessments but also according to assessments performed by the patients themselves.
Acknowledgments
The authors thank Susan DeRocco, PhD, of Complete Healthcare Communications Inc., and Martha Sensel, PhD, of MedProse, for assistance with drafting the manuscript on behalf of Amgen Inc., and Bojena Bitman of Amgen Inc. for biostatistical support.
Footnotes
-
Full Release Article. For details see Reprints/Permissions at jrheum.org
-
Supported by Immunex Corp., a wholly owned subsidiary of Amgen Inc., and by Wyeth Pharmaceuticals, which was acquired by Pfizer Inc. in October 2009.
- Accepted for publication February 10, 2010.
Free online via JRheum Full Release option








{kind=link}
{kind=link}
{kind=link}