Article Text

Abstract
Objective To elucidate the common and distinct clinical features of immune-mediated necrotising myopathy (IMNM), also known as necrotising autoimmune myopathy associated with autoantibodies against signal recognition particle (SRP) and 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR).
Methods We examined a cohort of 460 patients with idiopathic inflammatory myopathies (IIMs) through a muscle biopsy-oriented registration study in Japan. Study entry was strictly determined by the comprehensive histological assessment to exclude other neuromuscular disorders. Anti-SRP and anti-HMGCR antibodies were detected by RNA immunoprecipitation and ELISA, respectively.
Results Of 460 patients with IIM, we diagnosed 73 (16%) as having inclusion body myositis (IBM). Of 387 patients with IIMs other than IBM, the frequencies of anti-SRP and anti-HMGCR antibodies were 18% and 12%, respectively. One patient had both autoantibodies. Severe limb muscle weakness, neck weakness, dysphagia, respiratory insufficiency and muscle atrophy were more frequently observed in patients with anti-SRP antibodies than in those with anti-HMGCR antibodies. Serum creatine levels were markedly higher in the patients with autoantibodies than in those without. Histology was characterised by necrosis and regeneration of muscle fibres and was consistent with IMNM except in 1 HMGCR-positive IBM patient. Most patients were initially treated with corticosteroids; however, additional immunosuppressive drugs were required, especially in the patients with anti-SRP antibodies. Rates of unsatisfactory neurological outcome were similar in the 2 autoantibody groups.
Conclusions Anti-SRP antibodies are associated with severe neurological symptoms, more so than are anti-HMGCR antibodies. Although these autoantibodies are independent serological markers associated with IMNM, patients bearing either share common characteristics.
Statistics from Altmetric.com
Introduction
Inflammatory myopathies are a heterogeneous group of subacute, chronic, or acquired diseases of skeletal muscle.1 ,2 These myopathies involve skeletal muscle as well as many other organs, such as the lungs, heart, joints and skin. Inflammatory myopathies are the most commonly acquired immune-mediated and potentially treatable muscle diseases in children and adults. They are independently classified on the basis of distinct clinical or histological criteria. In fact, among patients with ‘clinical’ polymyositis, only 5% have ‘histological’ polymyositis.3 This discrepancy may cause misinterpretation in general physicians. The clinical phenotypes can be also defined by certain autoantibodies found in patients with inflammatory myopathies.4 ,5 It is of clinical importance to identify these autoantibodies because each autoantibody is closely associated with certain clinical manifestations.
Over the last decade, immune-mediated necrotising myopathy (IMNM), also known as necrotising autoimmune myopathy, has been recognised as a category of idiopathic inflammatory myopathy (IIM) characterised by necrosis in the absence of prominent inflammatory cells.6 ,7 IMNM is regarded as a different subtype from polymyositis, dermatomyositis and inclusion body myositis (IBM). The heterogeneous pathogenesis of necrotising myopathy includes autoantibody-mediated, drug-induced and paraneoplastic disease, along with overlap syndrome and viral infections.6–8 Autoantibodies against signal recognition particle (SRP) or 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR) are regarded as representative autoantibodies. Characteristic clinical manifestations associated with anti-SRP or anti-HMGCR antibodies have been described,9–13 but they have never been compared directly.
The purpose of the present study was to elucidate the common and distinct clinical features of IMNM associated with anti-SRP and anti-HMGCR antibodies among 460 patients with IIM through a muscle biopsy-oriented registration study in Japan.
Patients and methods
We collected samples for the ‘Integrated Diagnosis Project for Inflammatory Myopathies’ from October 2010 to December 2014.14 We received frozen muscle biopsy blocks and serum from patients with tentative diagnoses of inflammatory myopathies, from all over Japan. The following study entry criteria were used: (1) the patient was available for a muscle biopsy and could provide serum, accompanied by full clinical information, (2) the patient exhibited objective limb muscle weakness supported by electromyography and/or muscle MRI, (3) a diagnosis of inflammatory myopathies was made by a comprehensive histological examination and (4) the patient signed an informed consent agreement. This study was approved by the Institutional Review Boards of both the National Center of Neurology and Psychiatry, and Keio University.
We enrolled 460 patients (male:female=201:259; mean age at examination 55.2±20.8 years, range 2–84 years) with IIM by completely excluding other disorders, which included muscular dystrophies, dysferlinopathy, neurogenic muscular atrophy, clinically amyotrophic dermatomyositis, isolated neck extensor myopathy, glycogen storage disease, mitochondrial myopathy, infectious myositis, sarcoid myopathy and rhabdomyolysis, which were determined by clinical and histological assessment (see muscle histology section) (figure 1).

Study flow diagram. IIM, idiopathic inflammatory myopathy.
IBM was diagnosed by the identification of rimmed vacuoles with non-necrotic fibres invaded by mononuclear cells or increased major histocompatibility complex (MHC) class I expression.15 Polymyositis was diagnosed based on endomysial inflammation cell infiltrate surrounding or invading non-necrotic muscle fibres accompanied by ubiquitous MHC class I expression. Dermatomyositis was diagnosed by the identification of the presence of perifascicular atrophy. IMNM was diagnosed based on the observation of many necrotic fibres as the predominant abnormal histological feature without or with minimal lymphocytes infiltration. Other myopathies were regarded as non-specific myositis including perimysial and endomysial inflammatory cell infiltration.16
Clinical features
Each patient's clinical information was provided by his or her referring physician, who completed detailed charts including clinical course, neurological examination and laboratory findings. Skin manifestations including Gottron's rash and heliotrope sign were regarded as skin rash.1 ,2 We additionally asked for follow-up information of patients with anti-SRP or anti-HMGCR antibodies. Neurological outcomes were assessed using the modified Rankin Scale.10 Briefly, patients who responded to the treatment and returned to their jobs were defined as 0 or 1. Patients who responded partially to treatment and resumed most activities of daily living were defined as 2. Patients who responded minimally to the treatment and required support in daily activities were defined as 3–5.
Muscle histology
All of the clinical materials used in this study were obtained for diagnostic purposes with written informed consent. We processed the skeletal muscle tissue for pathological analysis at National Center of Neurology and Psychiatry.14 We stained serial 10 μm thick cryosections using a battery of histochemical methods including H&E, modified Gomori's trichrome, NADH-tetrazolium reductase, succinate dehydrogenase, cytochrome C oxidase, periodic acid-Schiff, acid phosphatase, oil red O, acetylcholinesterase, non-specific esterase, phosphorylase, phosphofructokinase, alkaline phosphatase, AMP deaminase, menadione-linked α-glycerophosphate dehydrogenase and myosin ATPase. To exclude a range of muscular dystrophies, we performed immunohistochemistry by using antibodies towards dystrophin, α-sarcoglycans to δ-sarcoglycans, α-dystroglycans and β-dystroglycans, dysferlin, caveolin-3, emerin, laminin α2, collagen VI, p62 and CD8. In addition to the histological studies, we performed mini-multiplex Western blotting using antidystrophin, antidysferlin, anticalpain 3, anti-α-sarcoglycan and antitelethonin.
We further analysed expression of MHC class I and II, and membrane attack complex (MAC), in patients with anti-SRP and anti-HMGCR antibodies. The patients whose muscle specimens did not include more than 300 fibres were excluded for this analysis. For MHC class I and II, we judged as positive only when the cytoplasm of non-necrotic/regenerating fibres were stained and negative if only sarcolemmata were stained. For MAC, the presence of at least one non-necrotic fibre with sarcolemmal deposits and that of at least one endomysial capillary with MAC deposition were regarded as positive.
Autoantibody detection
All sera were obtained before the treatment initiation. Frozen sera were stored at −30°C until use. Autoantibodies were detected by RNA immunoprecipitation at Keio University by researchers who were blind to the patients’ clinical and histological information.14 In addition, autoantibodies to HMGCR were measured using an ELISA test.17 These detection assays were performed in all samples, however, immunoblots or dermatomyositis-associated autoantibodies were not evaluated in all samples.
Statistical analyses
All analyses were performed using statistical analysis software (IBM/SPSS V.20, Armonk, New York, USA). Comparisons of relative frequencies were tested for significance using the χ2 test for 2×2 tables. Continuous variables were compared using the Mann-Whitney U-test. Values of p<0.05 were considered significant.
Results
Autoantibody profiles
Of 460 patients with IIM, we diagnosed 73 patients (16%) as having IBM. Frequencies of autoantibodies are shown in table 1. Anti-SRP antibodies were most frequently detected in 18% (69/387) of the patients with IIMs other than IBM. Subsequently, anti-HMGCR antibodies were found in 12% (46/387). In contrast, 13% (51/387) of patients possessed anti-aminoacyl transfer RNA synthetase (anti-ARS) antibodies. These three autoantibodies were found independently in all but one SRP/HMGCR dual-positive patient. In no patient with IBM except one, who had anti-HMGCR antibodies, were these three autoantibodies detected. IIMs except for IBM (n=387) were further divided into polymyositis (n=19), dermatomyositis (n=56), IMNM (n=177), antisynthetase syndrome (n=51) and non-specific myositis (n=84), since anti-synthetase syndrome is a clinically and pathologically distinct disease entity.18 ,19 Thus, anti-SRP and anti-HMGCR antibodies were found in 39% (69/177) and 26% (46/177) of the patients with IMNM.
Frequencies of autoantibodies
Clinical features
To evaluate the characteristics associated with anti-SRP and anti-HMGCR antibodies, we excluded two patients from the analysis, the one patient with both antibodies and the one patient with IBM with anti-HMGCR antibody. The resulting clinical and laboratory examinations compared anti-SRP (n=68) and anti-HMGCR (n=45) groups (table 2).
Comparison between anti-SRP and anti-HMGCR antibodies
There were no significant differences in the sex ratio and the age at examination. Fully 18% of the anti-HMGCR group had statin exposure, compared with 4% of the anti-SRP group (p=0.019). Patients with stain exposure tended to be older than those without in both groups (figure 2). Chronic progression of muscle weakness over 12 months, which mimicked muscular dystrophy, was observed equally in both groups.7 ,10 ,12
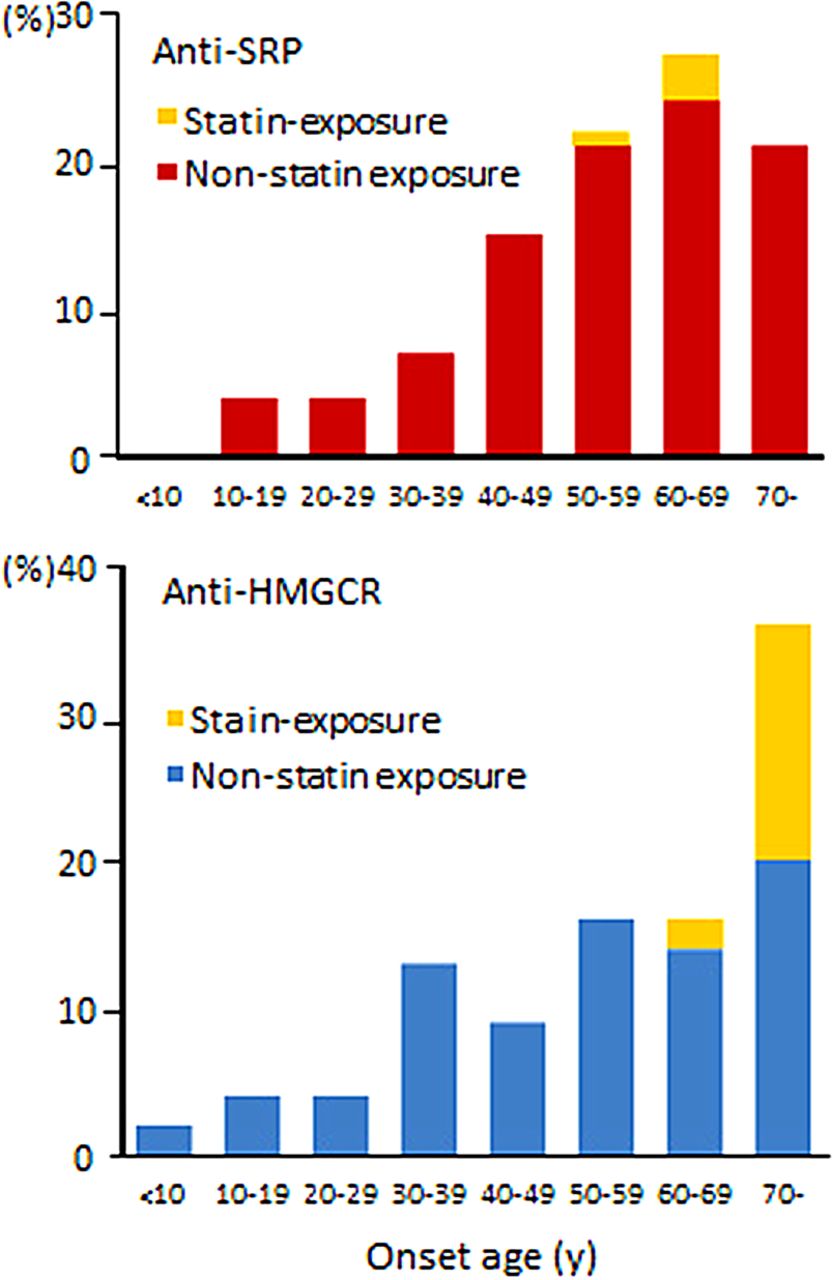
Distribution of onset age in 68 patients with anti-signal recognition particle (anti-SRP) antibodies and 45 patients with anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase (anti-HMGCR) antibodies.
Fifty-three patients (63%) of the anti-SRP group showed severe limb muscle weakness of grade ≤3/5 assessed by manual muscle strength (Medical Research Council scale grade), compared with 11 patients (24%) of the anti-HMGCR group (p<0.0001). Limb weakness usually affected legs more than arms. Laterality of limb weakness was seen in 18% of the anti-SRP group and in 13% of the anti-HMGCR group, respectively.
Neck weakness was found in 71% of the anti-SRP group, which was significantly higher than the corresponding value in the anti-HMGCR group (p=0.0055). Dysphagia was also highly detected in 68% of the anti-SRP group, compared with 44% of the anti-HMGCR group (p=0.014). Facial and cardiac involvements were infrequent in both groups. Respiratory insufficiency was observed in 12% of the anti-SRP group, but in 0% of the anti-HMGCR group (p=0.017). Muscle atrophy was seen in 68% of the anti-SRP group, significantly higher than the percentage among the anti-HMGCR group (p=0.014).
The frequencies of extramuscular manifestations including fever, skin rash, arthritis and Raynaud phenomenon, were low in both groups. Interstitial lung disease in chest CT, observed in 19% of the anti-SRP group, was tentatively attributed to respiratory insufficiency. With regard to associated disorders, the frequencies of cancer or concomitant rheumatic disease were not significantly different in the two groups.
Creatine kinase
Distribution of serum creatine kinase levels in all patients are shown in figure 3. Serum creatine levels were equally markedly elevated in the anti-SRP and anti-HMGCR groups. In contrast, the serum creatine kinase level of the patients with IBM was lower, at 632±458 IU/L. The serum creatine kinase level was significantly higher in the combined group of patients with anti-SRP and anti-HMGCR antibodies than in those without antibodies (6989±5034 vs 2764±6323 IU/L, p<0.0001). As indicated in table 2, there were no differences in serum creatine kinase level between the anti-SRP and anti-HMGCR groups. On the other hand, the range of serum creatine kinase in the ARS-positive patients was widely distributed: at an average of 4281±4981 IU/L.

Distribution of serum CK levels. CK, creatine kinase; IBM, inclusion body myositis; IIM, idiopathic inflammatory myopathy; HMGCR, 3-hydroxy-3-methylglutaryl-coenzyme A reductase; SRP, signal recognition particle.
Only one patient of the anti-SRP group and three patients of anti-HMGCR group had serum creatine kinase levels under 1000 IU/L. When the cut-off level of serum creatine kinase level was set as 1000 IU/L, the accuracy of the presence of anti-SRP or anti-HMGCR antibodies in total patients with IIM including IBM was calculated as 72%. In addition, 17 of 51 patients with anti-ARS antibodies had serum creatine kinase levels under 1000 IU/L.
In contrast, laboratory evaluation showed elevated C reactive protein and antinuclear antibody positivity (≥1:160) were not suggestive for the presence of anti-SRP or anti-HMGCR autoimmunity.
Histological features
Histological features of patients with anti-SRP and anti-HMGCR antibodies were characterised by necrosis and regeneration of muscle fibres, with no or little endomysial lymphocyte infiltration (figure 4A). Necrotic fibres are occasionally associated with macrophage infiltration (figure 4A, B).

Muscle histology. (A) Necrotic (black arrow) and regenerating (arrowhead) fibres. A white arrow indicates a necrotic fibre with macrophages (H&E stain). (B) Macrophages in necrotic fibres are highlighted by acid phosphatase. Scale bar: 50 μm.
Results of immunohistochemical analysis of MHC class I and II, and MAC, are represented in table 2. Increased expression of MHC class I on cytoplasm of non-necrotic/regenerating fibres was observed in 33 of 65 (51%) patients with anti-SRP antibodies and 21 of 43 (49%) patients with anti-HMGCR antibodies. The stainability tended to be faint as compared with other IIMs including polymyositis, dermatomyositis, and IBM. Perifascicular reinforcement was observed in two anti-SRP IMNM patients. No sample showed cytoplasmic upregulation of MHC class II. Deposition of MAC on sarcolemma of non-necrotic fibres was more commonly seen in patients with anti-HMGCR antibodies compared than in those with anti-SRP (65% vs 22%, p<0.0001). The fibres with sarcolemmal MAC deposits were usually small-sized, but occasionally the surfaces of apparently normal fibres were also covered with MAC deposits. Deposition of MAC on endomysial capillaries was observed in 17 of 65 (26%) patients with anti-SRP antibodies and 9 of 43 (21%) patients with anti-HMGCR antibodies. MAC deposition on capillaries was sparse and less intense as compared with that typically seen in dermatomyositis (data not shown).
Follow-up studies
Information regarding immunotherapy and neurological outcomes of 51 SRP-positive patients and 39 HMGCR-positive patients was available. Details of immunotherapy in the anti-SRR and anti-HMGCR groups were compared (table 3).
Comparison of treatment
Although corticosteroids were not administered in two patients due to severe side-effects, the other 88 patients were initially treated with corticosteroids. Some patients responded well to corticosteroids, without or with minimal neurological deficit. The frequency of corticosteroid monotherapy was higher in the anti-HMGCR group than in the anti-SRP group (31% vs 8%, p=0.0048).
In the other patients, although the immunotherapy resulted in a decrease of the patients’ creatine kinase levels, the recovery of muscle weakness was insufficient and they needed additional immunotherapy. Administration of intravenous immunoglobulin or intravenous methylprednisolone pulse therapy was repeatedly performed in both groups. To avoid side effects of corticosteroids, other additional immunosuppressive drugs were utilised. Among these drugs, tacrolimus was preferentially selected more often in the anti-SRP group than in the anti-HMGCR group (45% vs 13%, p=0.001). Plasma exchange was also performed in two treatment-resistant patients with anti-SRP antibodies.
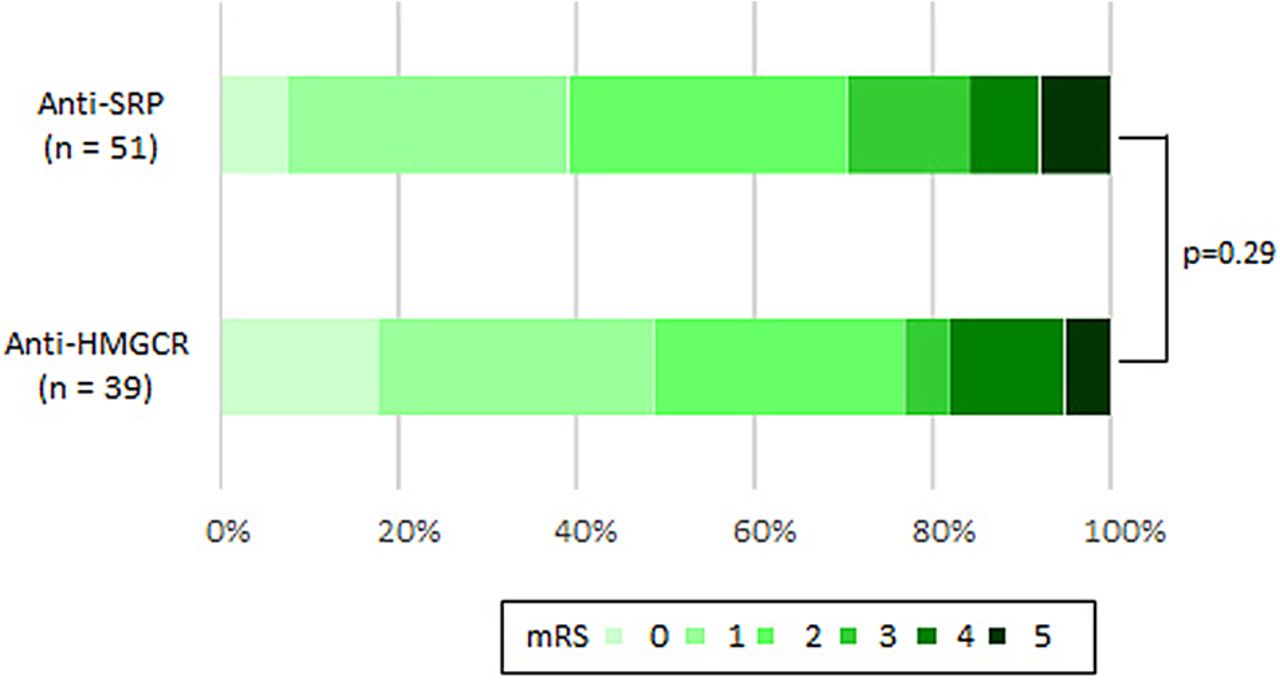
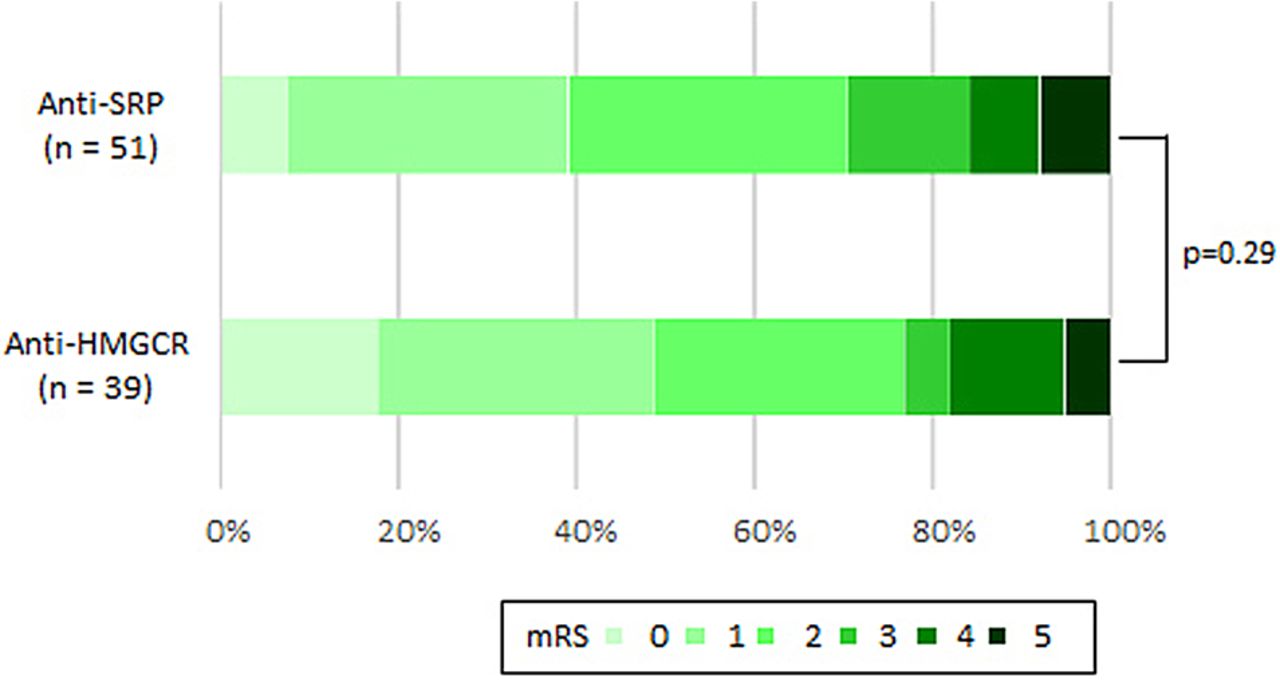
The patients’ neurological outcomes assessed using the modified Rankin Scale clearly showed incomplete improvement (figure 5). There were no differences in neurological deficits between the anti-SRP and anti-HMGCR groups. Fifteen patients (29%) of the anti-SRP group and nine (23%) of the anti-HMGCR group suffered from difficulties in their daily living graded as modified Rankin Scale scores 3–5.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Neurological outcomes using mRS. HMGCR, 3-hydroxy-3-methylglutaryl-coenzyme A reductase; mRS, modified Rankin Scale; SRP, signal recognition particle.
Discussion
The present study's finding can be summarised as follows: (1) the frequencies of anti-SRP and anti-HMGCR antibodies in patients with IIM except IBM were 18% and 12%, respectively; (2) severe limb muscle weakness, neck muscle weakness, dysphagia, respiratory insufficiency and muscle atrophy were more frequently observed in the patients with anti-SRP antibodies than in those with anti-HMGCR antibodies and (3) the rates of unfavourable neurological outcome were similar in the two groups of patients.
Previous studies reported that the frequencies of anti-SRP or anti-HMGCR antibodies were 5–10% in patients with polymyositis and dermatomyositis.4 ,5 ,9–13 The frequencies of anti-SRP and anti-HMGCR antibodies in the present study were higher than in previous studies. The reason is due to differences in study populations. The strength of our study is that study entry was strictly determined by comprehensive histological assessment in combination with Western blot—or, occasionally, genetic analysis—to fully exclude neuromuscular disorders other than IIM. In addition, the registration into the present project was made mostly by neurologists or neuromuscular specialists who manage patients with severe muscle weakness, which is more common in IMNM than in classical polymyositis and dermatomyositis. On the other hand, the frequency of anti-ARS antibodies was lower in this cohort than in those in previous studies. Anti-ARS antibodies, but not anti-SRP and anti-HMGCR, can be routinely examined in Japan. The reason for the lower frequency is that the patients proven to have anti-ARS antibodies are encouraged to avoid muscle biopsy.
The detection of autoantibodies such as anti-ARS antibodies and dermatomyositis-associated autoantibodies is useful for diagnosis, determination of treatment strategy and prediction of outcomes.4 ,5 In a like manner, anti-SRP and anti-HMGCR antibodies are indispensable markers for IMNM, and also help differentiate IMNM from muscular dystrophy.7 ,10 ,12 ,20 Serum creatine kinase levels were markedly increased in patients with these autoantibodies. These autoantibodies should be evaluated early for patients with myopathy especially when serum creatine kinase levels are elevated to >1000 IU/L.
We reported that the neurological outcome of SRP-positive patients was unsatisfactory regardless of the combination of immunosuppressive agents.21 In addition, the analyses revealed that younger disease onset was the most significant factor associated with poor neurological outcome. Anti-HMGCR antibodies were initially reported as a marker of statin-associated myopathy.11 This subset usually affects older people and the response to treatment is usually favourable. Subsequently, as several independent groups have investigated the antibodies, the ratio of statin-exposed patients among anti-HMGCR antibody-positive patients has declined from 63–73% to 15–44%, the latter consistent with the result of the present study (18%).11–13 ,17 ,22 The lower frequencies may suggest that production of anti-HMGCR antibodies is not directly related to statin treatment. Although we considered the patients receiving statin medication at symptom onset as statin-associated, the possibility of coincidence was not fully excluded. In fact, statin treatment is regarded as a risk factor of IMNM as well as of cancer and connective tissue disease.8
Furthermore, since the present comparison of clinical profiles of the patients with anti-SRP or anti-HMGCR IMNM in the same cohort was performed, anti-SRP antibodies have been shown to be more associated with severe limb muscle weakness and frequent involvement of neck weakness, dysphagia, respiratory insufficiency and muscle atrophy.
While an obvious difference could be recognised on neither conventional histochemistry nor on immunohistochemistry of MHC class I and II, we noted that sarcolemmal MAC deposition was more commonly seen in anti-HMGCR IMNM patients than in those with anti-SRP. In prior studies, sarcolemmal MAC deposition has been raised as a common pathological feature of anti-HMGCR IMNM.12 ,23 ,24 Our study suggests that the MAC deposition could be a characteristic feature of anti-HMGCR NAM, however, these findings may be non-specific.
We are aware of a few study limitations. First, one consequence of our integrated diagnosis project was that results of autoantibodies were proven at different timings. We reported the positivity of anti-SRP antibodies to each physician several months after the start of treatment. The presence of anti-SRP antibodies may be useful for physicians considering more aggressive immunotherapy when their patients are not responding to corticosteroids. On the other hand, we only established the detection system of anti-HMGCR antibodies in 2014.17 Thus, positivity of anti-HMGCR was not taken into consideration in immunotherapy in our study. This study has demonstrated that anti-HMGCR IMNM shows overall equivalence to anti-SRP IMNM in terms of neurological outcome. However, if the results of anti-HMGCR antibodies were available before treatment initiation, neurological outcomes might have been more favourable.
Second, duration of therapeutic regimens and time of therapy initiation were varied in the present study. In addition, there was no consensus on the regimens of immunotherapy. Therefore, we did not mention which agents were more beneficial to patients with anti-SRP/HMGCR IMNM. Intravenous immunoglobulin was preferentially selected, as it has been approved, by the Japanese government, for the treatment of inflammatory myopathies. Among steroid-sparing agents, mycophenolate mofetil, methotrexate and azathioprine, are generally administrated in the USA and in European countries.8 ,12 In contrast, tacrolimus is frequently chosen in Japan. This may be because physicians are familiar with the use of tacrolimus in the treatment of other autoimmune diseases. In this regard, rituximab is also a good candidate for treatment-resistant patients.25 A double-blind and placebo-controlled study is required to decide the best regimens of immunotherapy for patients with anti-SRP/HMGCR IMNM. Finally, it is important to confirm the decreased levels of anti-SRP and anti-HMGCR antibody titres over time under treatment.9 ,26
In conclusion, anti-SRP antibodies were associated with a particular phenotype including severe muscle weakness and atrophy, compared with anti-HMGCR antibodies.
Acknowledgments
The authors thank the physicians who provided muscle biopsy, serum samples, and detailed clinical information. They are grateful to Ms Kaoru Tatezawa and Ms Kazu Iwasawa (Department of Neuromuscular Research, National Institute of Neuroscience, and Department of Genome Medicine Development, Medical Genome Center, National Center of Neurology and Psychiatry) for their excellent technical support.
References
Footnotes
YW and AU contributed equally to this study.
Contributors YW, AU, SS and IN were involved in the drafting of the manuscript or revising it carefully for important intellectual content, and all the authors have read and approved the final version of the manuscript. SS had full access to all of the data in the study, and takes responsibility for the integrity of the data and the accuracy of the data analysis. AU, SS and NS contributed to the study concept and design. YW, AU, SS, JN, KH and KT contributed to the acquisition of data. AU, SS, JN, SS and IN contributed to the analysis and interpretation of the data.
Funding The work of SS was supported by the Japanese Ministry of Education, Science, Sports and Culture (number 26461298), and Health and Labor Sciences Research Grant on Intractable Diseases (neuroimmunological diseases) from the Ministry of Health, Labor and Welfare of Japan. The work of IN was supported by a Grant-in-Aid for Scientific Research from MEXT (number 24390227).
Competing interests None declared.
Patient consent Obtained.
Ethics approval The Institutional Review Boards of both the National Center of Neurology and Psychiatry, and Keio University.
Provenance and peer review Not commissioned; externally peer reviewed.