Article Text

Abstract
Background: Most of the available documentation in the literature on ocular involvement in localised scleroderma (LS) are descriptions of single cases in adult patients. This article reports the frequency and specific features of ocular involvement in a large cohort of children with juvenile LS (JLS).
Methods: Data from a large, multi-centre, multinational study of children with LS were used to collect and analyse specific information on ocular involvement.
Results: 24 out of 750 patients (3.2%) revealed a significant ocular involvement. 16 were female and 8 male. 16 patients (66.7%) had scleroderma “en coup de sabre” (ECDS) of the face, 5 (20.8%) had the linear subtype, 2 (8.3%) had generalised morphea (GM) and one (4.2%) had plaque morphea (PM). Of the 24 patients with eye involvement, 10 patients (41.7%) reported adnexa (eyelids and eyelashes) abnormalities, 7 (29.2%) anterior segment inflammation (5 anterior uveitis, 2 episcleritis) and 3 central nervous system-related abnormalities. 4 patients presented single findings such as paralytic strabismus (1), pseudopapilloedema (1) and refractive errors (2). Other extracutaneous manifestations were detected in a significantly higher number of patients with ocular involvement and were mostly neurological.
Conclusion: Ocular abnormalities are not unusual in patients with JLS, especially in the ECDS subtype. They are frequently associated with other internal organ involvement, particularly the central nervous system (CNS). Careful ophthalmic monitoring is recommended for every patient with JLS, but is mandatory in those with skin lesions on the face and/or concomitant CNS involvement.
Statistics from Altmetric.com
Juvenile localised scleroderma (JLS) is the most frequent subtype of scleroderma in children. It involves mainly the skin and includes four subtypes: plaque morphea (PM), generalised morphea (GM), deep morphea (DM) and linear scleroderma.
Each of these subtypes can involve the face and particularly the orbit. Linear scleroderma of the face, also known as “en coup de sabre” because it resembles the strike of a sword,1 affects the frontoparietal area of the head and often involves the ocular adnexa and even the eye.
The data on ocular involvement in localised scleroderma (LS) consist essentially of single case reports of adult patients with a wide variability of manifestations.2–5 Very few reports deal with this complication in children.6 7
The aim of the present study was to evaluate the frequency and the characteristics of ocular involvement in a large cohort of children with JLS from a multi-centre international data collection.
METHODS
As part of an international project on classification of subtypes of JLS, a data collection form was distributed to 270 paediatric rheumatology and dermatology centres in Europe (166), the USA (42), South America (28), Asia (30), Australia (2) and Africa (2). The aim was to gather information on the following demographic, epidemiological, clinical and laboratory characteristic features of children with JLS:
Gender.
Age at diagnosis.
Clinical subtype according to the Mayo Clinic classification criteria.1 This classification distinguishes five groups of localised scleroderma:
plaque morphea (PM)
generalised morphea (GM)
bullous morphea
linear scleroderma including the head–face subtype en coup de sabre (ECDS)
deep morphea (DM).
Clinical description of the ocular involvement including the residual damage at the last evaluation.
The presence of other internal organ involvement, such as the neurological, cardiac, musculoskeletal, respiratory and vascular systems.
Autoantibody profile: antinuclear antibodies (ANA), anticardiolipin antibodies (ACL) and rheumatoid factor (RF). Abnormal values were referred to the normal range of laboratory standards of each participating centre.
Since all clinical information was anonymously collected from the patients’ charts, Institutional Review Board approval was required only from a minority of centres, mainly in the USA.
Statistical analysis
Demographic, clinical and laboratory characteristics of the patients with ocular involvement were analysed by descriptive statistics. Differences between the two groups of patients, with and without ocular involvement, were analysed by the t test, Χ2 test or Fisher’s exact test, as appropriate. A value of p<0.05 was considered significant. All analyses were performed using the SAS statistical software (Version 8.0, SAS Institute Inc., USA).
RESULTS
The clinical information of 750 patients with JLS who were followed at 69 paediatric rheumatology and dermatology centres was reviewed.
Twenty-four patients (3.2%) had a significant ocular involvement and their clinical characteristics are summarised in tables 1 and 2. The female:male ratio was 1.9:1, and the mean age at the onset of scleroderma was 5 years and 6 months (range 2 months–14.3 years). Follow-up ranged between 10 months and 22 years (mean 6.6 years).
Sixteen patients (66.7%) had the ECDS variety and 5 (20.8%) had the linear subtype involving the limbs and the trunk. Two patients presented with GM and one with PM.
In the whole group of 113 patients with the ECDS subtype, ocular involvement was present in 16 (14.2%), a significantly higher frequency than in the other subtypes (3.9% in the GM, 1.3% in the linear subtype, 0.8% in the PM).
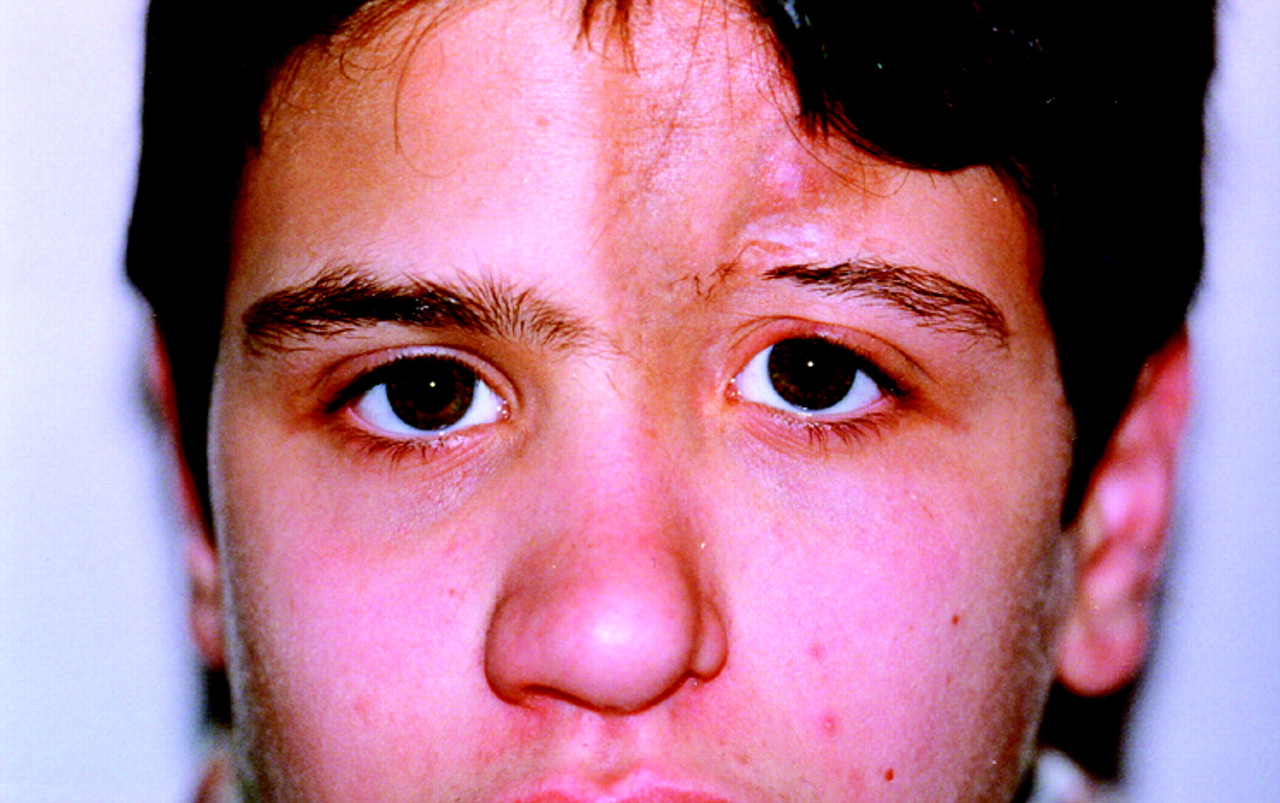
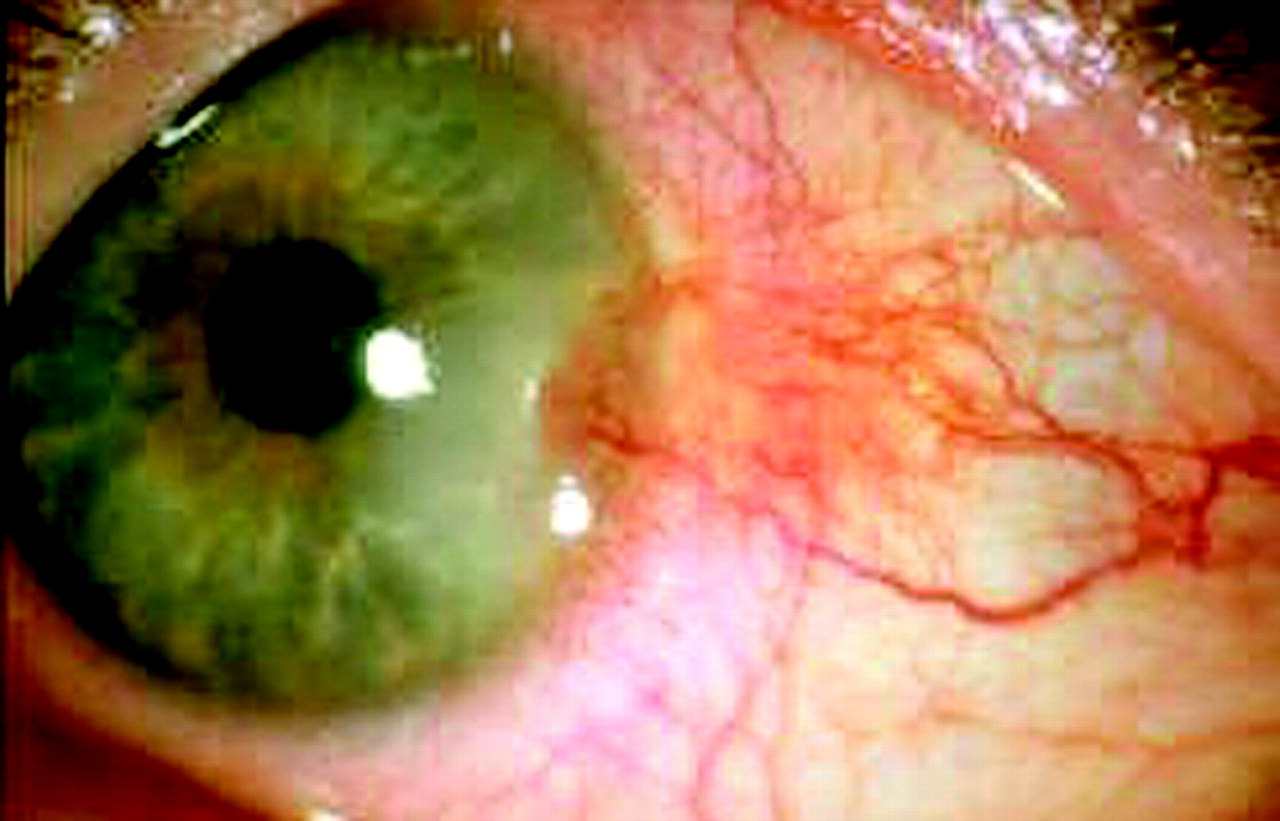
Several ocular findings have been reported. Adnexal abnormalities involving eyelids and eyelashes or lachrymal glands were the most frequent findings, reported in 10 patients (41.7%), eight with ECDS and two with GM (fig 1). Anterior segment inflammation (ASI) was present in seven patients (29.2%). Anterior uveitis (AU) was found in five patients and was complicated by secondary glaucoma in one. In all, AU was asymptomatic and unilateral. Two had episcleritis, in one associated with the linear subtype (fig 2), in another with ECDS.

{kind=link}
{kind=link}
Three patients (12.5%) had CNS-related abnormalities. Pupillary mydriasis was the ocular manifestation concomitant with CNS involvement in two patients with ECDS. One presented with epilepsy and the other with pseudotumour cerebri and left orbital myositis. Another patient with ECDS presented a complicated picture consisting of enophthalmos, anisocoria, partial iris atrophy, stellate neuroretinitis and retinal teleangiectasiae. Interestingly, this patient presented with concomitant CNS abnormalities of MRI of the thalamus and cerebellar hemisphere.
There were four patients with miscellaneous eye findings. One patient with ECDS presented with a paralytic strabismus of cranial nerve VI, without other neurological or MRI abnormalities. Pseudopapilloedema was present in one patient with the linear subtype. Refractive errors such as myopia and astigmatism were reported in two patients and, since they are common problems in children, should probably be considered incidental findings.
In 10 patients (41.7%), a concomitant involvement of other internal organs was present. Six had neurological involvement (25.0%), consisting of epilepsy (2), peripheral neuropathy (1), MRI abnormalities (2) and pseudotumour cerebri (1). Arthritis was present in 2 (8.3%); aortic insufficiency, abnormal pulmonary function tests and Raynaud phenomenon were present in three other individual cases, respectively. As far as serological features, ANA were found to be positive in 12 patients (50.0%) but there was no significant association with a particular ophthalmological finding. RF was positive in two patients, both with concomitant osteoarticular involvement.
Comparing patients with and without ocular involvement (table 2), we found that the age at disease onset was significantly lower in the first group. As expected, ECDS was the prevalent subtype, being present in 66.7% of patients with ocular involvement (p<0.001). Indeed, patients with ocular involvement also have a higher prevalence of internal organ involvement: 45.8% versus 21.6% in the non ocular-group (p<0.01). In particular, neurological involvement was significantly associated with ocular abnormalities (p<0.005).
DISCUSSION
Ocular involvement is not common in the whole group of children with JLS, being present in just 3.2% of the patients. However, ocular abnormalities were found in 14% of the patients with ECDS.
This is the largest series of JLS patients with ocular involvement. Interestingly, these patients are significantly younger at the onset of the disease and the ocular involvement appears quite early in the disease course. The ocular abnormalities found in our series can be categorised into four groups of conditions.
In the first group are gathered the consequences of the fibrotic involvement of the eyelids and eyelashes or lachrymal glands with dry eye. This complication, present in 10 patients, was the most frequent. As reported in adult patients, these abnormalities and dry eyes may be complicated by the inability to blink and sometimes ectropion with risk for exposure keratopathy.11 12 In this group of patients the lesion was quite evident and the diagnosis was early and appropriate.
The involvement of the anterior segment of the eye was the second most frequent condition, being present in seven patients (29.2%). AU, found in five cases, was the most relevant finding. As in juvenile idiopathic arthritis,14 this condition represents a manifestation of a systemic autoimmune process. Anterior uveitis can be completely unrelated to the site of the cutaneous involvement, as happened in three patients, and is asymptomatic. For this reason, a periodic ophthalmic monitoring should be performed, especially in this subgroup of patients, to avoid complications such as glaucoma, reported in patient number 14. Episcleritis, found in two patients, was probably the expression of a subconjunctival inflammatory-fibrotic lesion similar to the cutaneous ones (fig 2). Unlike other connective tissue diseases, these lesions were quite steroid-resistant, probably due to their prevalent fibrotic component.
The third group includes the so-called CNS-related abnormalities. In these patients the ocular lesions were associated with varieties of neurological abnormalities. Therefore, in patients with dilated pupil and complex ocular abnormalities (as in patient no. 20), appropriate CNS imaging studies should be performed. Conversely, once neurological symptoms or CNS imaging alterations are present, a complete ophthalmic evaluation is mandatory. We may speculate a possible causative relationship between these ocular abnormalities and the CNS involvement, although no clear pathophysiological mechanisms have been found.
The last group of conditions include a variety of conditions such as paralytic strabismus, pseudopapilloedema and refractive errors, in which, again, a causative relationship with scleroderma is difficult to prove. A prospective collection of data on the ophthalmic involvement in JLS patients is needed to clarify the real meaning of these alterations.
In previous studies, ocular involvement has been described in 10% of adult patients with localised scleroderma2 and in one third of those with hemifacial atrophy.4 The reported alterations, as in our study, may involve adnexa structures,3 the anterior or posterior segments of the eye4 6 7 and the CNS.5 8–10 The involvement of the extrinsic eye muscles, as in one of our patients (no.19), is extremely rare but well described by both CT and MRI4 13 and should be suspected in presence of pain on eye movement, diplopia and strabismus.
Another important finding in our study was the high prevalence of a concomitant involvement of other internal organs detected in almost half of the patients with ocular manifestations (table 2). The most common association was with CNS abnormalities, underlying the close relationship between these two organ systems, as reported in adults.5 These observations, together with the fact that ocular lesions are seen even in subtypes of scleroderma without facial involvement, may support the concept that JLS is a systemic disorder.
CONCLUSION
Ocular abnormalities are not rare in patients with JLS involving the face as in ECDS. They are frequently associated with other internal organ involvement, especially the CNS. A careful ophthalmic monitoring is recommended for every patient with JLS, but is mandatory in those with skin lesions on the face and/or concomitant CNS involvement.
In particular, since the disease evolves rapidly during the first 2–3 years, we suggest an ophthalmic screening every 3–4 months for the first 3 years and then only in case of relapse.
REFERENCES
Footnotes
Competing interests: None declared.
- Abbreviations:
- ACL
anticardiolipin antibodies
- ANA
antinuclear antibodies
- AU
anterior uveitis
- CNS
central nervous system
- DM
deep morphea
- ECDS
en coup de sabre
- GM
generalised morphea
- JLS
juvenile localised scleroderma
- LS
localised scleroderma
- PM
plaque morphea
- RF
rheumatoid factor