Article Text

Abstract
Objectives Vitamin D deficiency is widespread and has been associated with many chronic diseases, including autoimmune disorders. A study was undertaken to explore the impact of low vitamin D levels on autoantibody production in healthy individuals, as well as B cell hyperactivity and interferon α (IFNα) activity in patients with systemic lupus erythematosus (SLE).
Methods Serum samples from 32 European American female patients with SLE and 32 matched controls were tested for 25-hydroxyvitamin D (25(OH)D) levels, lupus-associated autoantibodies and serum IFNα activity. Isolated peripheral blood mononuclear cells were tested for intracellular phospho-ERK 1/2 as a measure of B cell activation status.
Results Vitamin D deficiency (25(OH)D <20 ng/ml) was significantly more frequent among patients with SLE (n=32, 69%) and antinuclear antibody (ANA)-positive controls (n=14, 71%) compared with ANA-negative controls (n=18, 22%) (OR 7.7, 95% CI 2.0 to 29.4, p=0.003 and OR 8.8, 95% CI 1.8 to 43.6, p=0.011, respectively). Patients with high B cell activation had lower mean (SD) 25(OH)D levels than patients with low B cell activation (17.2 (5.1) vs 24.2 (3.9) ng/ml; p=0.009). Patients with vitamin D deficiency also had higher mean (SD) serum IFNα activity than patients without vitamin D deficiency (3.5 (6.6) vs 0.3 (0.3); p=0.02).
Conclusions The observation that ANA-positive healthy controls are significantly more likely to be deficient in vitamin D than ANA-negative healthy controls, together with the finding that vitamin D deficiency is associated with certain immune abnormalities in SLE, suggests that vitamin D plays an important role in autoantibody production and SLE pathogenesis.
Statistics from Altmetric.com
Vitamin D deficiency has been associated with several autoimmune diseases including multiple sclerosis (MS), rheumatoid arthritis (RA), type 1 diabetes mellitus, inflammatory bowel disease (IBD), mixed connective tissue disease, autoimmune thyroid disease, scleroderma and systemic lupus erythematosus (SLE).1,–,9 Vitamin D levels are lower in individuals with undifferentiated connective tissue disease than in controls, and lower in patients who progressed to well-established connective tissue diseases than in those whose did not progress.10
Vitamin D supplementation has been shown to improve disease in murine models of MS, RA, type 1 diabetes mellitus, IBD and SLE.11 Addition of vitamin D and its synthetic analogues to murine models of SLE have resulted in improved dermatological disease, reduced proteinuria and increased survival.1 SLE is a complex heterogeneous autoimmune disorder arising from genetic predisposition and environmental risks.12 13 Vitamin D deficiency has been found in approximately two-thirds of patients with SLE, with approximately one-fifth of patients having severe deficiency (<10 ng/ml).2 Additionally, serum vitamin D levels have been shown to correlate inversely with disease activity.14,–,16
Vitamin D has modulatory effects on B lymphocytes and Ig production. Many immune cells contain vitamin D receptors—including monocytes, macrophages, dendritic cells and activated T and B cells—and these immune cells possess the enzymatic machinery (1α-hydroxylase, CYP27B1) necessary to convert vitamin D into its active form.17 Isolated peripheral blood mononuclear cells (PBMCs) from patients with SLE incubated with 1,25(OH)2D or its synthetic analogues significantly reduced cellular proliferation, as well as polyclonal and anti-dsDNA Ig production.18 1,25(OH)2D has also been shown to induce apoptosis in activated B cells and to inhibit the generation of plasma cells and postswitch memory B cells, as well as induce regulatory T cells.19 20 The importance of vitamin D in innate immunity has been highlighted by studies demonstrating that monocyte/macrophage responses to bacterial infections via Toll-like receptors (TLRs) are potentially stimulated by 25-hydroxyvitamin D (25(OH)D) following localised induction of both vitamin D receptor (VDR) and 1α-hydroxylase.17
Interferon α (IFNα) has been shown to be a key cytokine in the pathogenesis of SLE. Numerous studies have confirmed the association between raised IFNα levels and increased disease activity in SLE.21 Strong evidence for the pathogenic role of IFNα in SLE comes from clinical studies showing treatment with recombinant IFNα for malignancies or hepatitis causes de novo SLE, with resolution after discontinuation of treatment.21 Induction of IFNα from stimulation of TLRs by nucleic acid-containing immune complexes is thought to be one of the mechanisms by which patients with SLE have increased IFNα activity.21 Supporting this hypothesis is evidence that activation of the IFNα pathway is associated with the presence of autoantibodies directed against DNA and RNA binding proteins.22 23
While in vitro studies have demonstrated a suppressive action of vitamin D on Ig production and the IFN signature,18 24 25 an association between vitamin D status in patients with SLE and these disease features has not been previously reported. We examined the prevalence of vitamin D deficiency in antinuclear antibody (ANA)-positive healthy individuals in comparison with ANA-negative healthy individuals and patients with SLE. The relationship between vitamin D status and the interrelated pathways involving B cell activation, autoantibody production and IFNα activity in SLE and healthy individuals was also evaluated.
Methods
Study population
Experiments were performed in accordance with the Helsinki Declaration and approved by the Institutional Review Boards at the Oklahoma Medical Research Foundation and the University of Oklahoma Health Sciences Center. All the study participants provided written informed consent prior to enrolment. All 32 female patients with SLE met at least four of the American College of Rheumatology (ACR) classification criteria.26 27 Additionally, healthy individuals were recruited to provide control samples and were matched to the patients based on age (±5 years), race and sex. Only European American females who are Oklahoma residents were included in the study in order to minimise variations in 25(OH)D levels due to race, sex and latitude.
Data collection
Demographic information included sex, age and self-reported race. Data were extracted from the medical records of the patients with SLE for ACR classification criteria, age of diagnosis and medication use. Peripheral blood was collected from each participant. Serum samples and plasma were isolated and stored at −20°C until further use. Patients with SLE were evaluated for disease activity by the Safety of Estrogen in Lupus Erythematosus National Assessment (SELENA)-modified SLE disease activity index (SELENA-SLEDAI), physician global assessment (PGA) and systemic lupus activity measure (SLAM). Controls completed the Connective Tissue Disease Screening Questionnaire (CSQ).28
25(OH)D levels
Plasma 25(OH)D levels were determined in duplicate using an enzyme immunoassay kit (Immunodiagnostic Systems, Scottsdale, Arizona, USA) according to the manufacturer's instructions. For this study, vitamin D deficiency was defined as ≤20 ng/ml.29 Vitamin D levels were determined from plasma samples obtained on the same date as the blood specimens procured to measure autoantibodies, serum IFNα activity and B cell activation.
Lupus-associated autoantibodies
ANAs were detected using an HEp-2 indirect immunofluorescent assay (INOVA Diagnostics, San Diego, California, USA) according to the manufacturer's instructions. Anti-double stranded DNA (dsDNA) antibodies were detected using a Crithidia luciliae indirect immunofluorescent assay (INOVA Diagnostics) according to the manufacturer's instructions. Detection of ANA at a dilution of 1:120 or greater and anti-dsDNA at a dilution of 1:30 or greater was considered a positive result. ELISAs were used to evaluate serum for antibodies to Sm, nuclear ribonucleoprotein (nRNP), Ro, La, ribosomal P (ribo P) and cardiolipin, as previously described.30 Samples were run in duplicate and normalised to a known positive control.
B cell activity markers
PBMCs (1×106) isolated by Ficoll-Paque gradients of fresh acid-citrate dextrose-anticoagulated blood were incubated at 37°C for 15 min and then washed and permeabilised for 10 min with BD Phosflow Perm/Wash Buffer I (BD Biosciences, San Jose, California, USA). After permeabilisation, cells were incubated with anti-CD79a-PE (BD Biosciences) and anti-phospho-Erk (pERK1/2) (Cell Signaling Technology, Danvers, Massachusetts, USA). The cells were then washed, stained with FITC goat anti-rabbit IgG (Jackson Immunoresearch, West Grove, Pennsylvania, USA) and analysed using FACS Calibur (BD Biosciences). Representative flow cytometry gating and staining are shown in figure 1 in the online supplement. Median fluorescent intensity values for the levels of pERK1/2 were used on CD79a-gated B cells to rank and normalise the data.
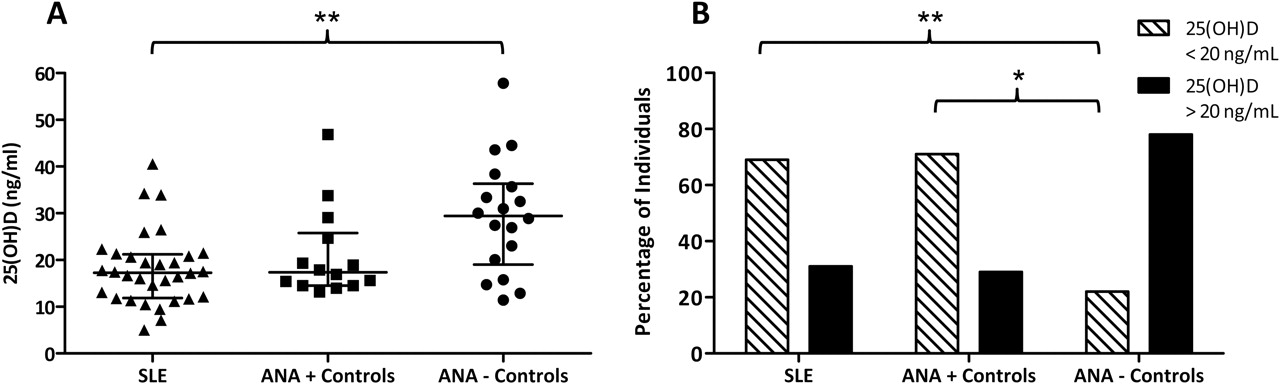
Antinuclear antibody (ANA)-positive healthy individuals and patients with systemic lupus erythematosus (SLE) are more likely to be deficient in vitamin D. (A) Median (IQR) 25-hydroxyvitamin D levels were 17.3 (11.9–21.2) ng/ml in patients with SLE (n=32), 17.4 (14.5–25.8) ng/ml in ANA-positive controls (n=14) and 29.4 (19.0–36.3) ng/ml in ANA-negative controls (n=18). Bars indicate IQR. **p<0.01, Kruskal–Wallis test with Dunn's multiple comparison. (B) Patients with SLE and ANA-positive controls were more likely to be vitamin D deficient (69% and 71%, respectively) than ANA-negative controls (22%). **p=0.003, *p=0.011 (Fisher exact test).
Serum IFNα activity
The reporter cell assay for serum IFNα activity has previously been described in detail.31 Briefly, reporter cells were used to measure the ability of patient serum to upregulate IFN-induced gene expression. The reporter cells (Wistar Institute, Susan Hayflick cells, ATCC No. CCL-25, American Type Culture Collection, Manassas, Virginia, USA) were cultured with 50% patient serum for 6 h and then lysed. mRNA was purified from cell lysates and cDNA was made from total cellular mRNA. cDNA was then quantified using real-time PCR. Forward and reverse primers for the genes MX1, PKR and IFIT, which are known to be highly and specifically induced by IFNα, were used in the reaction.32 Background gene expression was controlled by amplifying glyceraldehyde 3-phosphate dehydrogenase. IFNα activity values reported represent the number of SD above the mean of healthy donors (n=141). In this study, IFNα activity values >1 are considered high.
Statistical analysis
Mean 25(OH)D levels were compared using the Kruskal–Wallis test with the Dunn multiple comparisons post-test. Categorical data were analysed using the Fisher exact test. Continuous variables were analysed using an unpaired t test. Welch's correction was used in instances of unequal variance. Log-transformation was done to normalise serum IFNα activity measures. Pearson correlation was done to assess the relationship between pERK1/2 and 25(OH)D values. All statistical analyses were carried out with GraphPad Prism Version 5.01 (GraphPad Software, San Diego, California, USA; http://www.graphpad.com).
Results
Healthy European American ANA-positive individuals and patients with SLE are more likely to have vitamin D deficiency
25(OH)D levels were measured in 32 European American female patients with SLE and in 32 healthy matched controls. Fourteen of the controls were determined to be ANA-positive. The SLE patient group had a median 25(OH)D level of 17.3 (IQR 11.9–21.2) ng/ml (figure 1A). Interestingly, the 14 controls who were ANA-positive had a median 25(OH)D level of 17.4 (14.5–25.8) ng/ml, which was not statistically different from the patients with SLE (Kruskal–Wallis test with Dunn's post-test). In contrast, the 18 controls who were ANA-negative had significantly higher 25(OH)D levels than the patients with SLE (p<0.01, figure 1A).
The mean age of the 14 ANA-positive controls was significantly higher than the ANA-negative controls (54.1 vs 42.8 years; p=0.029, unpaired t test). However, in a logistic regression model with vitamin D status as the outcome and ANA positivity and age as the predictors, age did not predict vitamin D status (p=0.331) while ANA positivity remained a significant predictor (p=0.025). ANA-positive and ANA-negative controls had no significant difference in CSQ scores (median 1.0 (IQR 0.8–2.5) vs 1.0 (IQR 0.0–2.0); p=0.41, Mann–Whitney test). In addition, there was no difference in body mass index (BMI) values between ANA-positive and ANA-negative controls (26.1 vs 28.4 kg/m2; p=0.38, unpaired t test).
Furthermore, when categorised into vitamin D-deficient (25(OH)D <20 ng/ml) or non-deficient (25(OH)D >20 ng/ml), patients with SLE and ANA-positive controls were more likely to be vitamin D deficient (69% and 71%, respectively) than ANA-negative controls (22% deficient) (OR 7.7, 95% CI 2.0 to 29.4, p=0.003 and OR 8.8, 95% CI 1.8 to 43.6, p=0.011, respectively, Fisher exact test, figure 1B). Patients with deficient levels of 25(OH)D (n=22) had no statistically significant differences among specific ACR classification criteria, disease activity scores (SLEDAI, SLAM, PGA), BMI or medication use than patients with non-deficient levels of 25(OH)D (n=10). The median (IQR) prednisone dosage for vitamin D-deficient patients was 3 (0–5) mg/day compared with 0 (0–5) mg/day for those not deficient in vitamin D (p=0.507, Mann–Whitney test). Twenty-three per cent of vitamin D-deficient patients versus 20% of non-deficient patients were receiving treatment with azathioprine (p=1.000), 77% versus 60% were taking hydroxychloroquine (p=0.407), 23% versus 10% were receiving mycophenolate mofetil (p=0.637) and 23% versus 10% were being treated with methotrexate (p=0.637) (Fisher exact test). In addition, the percentage of vitamin D-deficient patients who were considered to be immunosuppressed (defined as receiving >10 mg/day prednisone, azathioprine, methorexate or mycophenolate mofetil) was not significantly greater than the percentage of immunosuppressed non-deficient patients (64% vs 40%; p=0.267, Fisher exact test). Vitamin D-deficient patients had a higher mean number of ACR classification criteria (6.4 vs 5.3; p=0.026, unpaired t test).
Increased B cell activation is associated with decreased 25(OH)D in SLE
An increase in B cell activation as measured by pERK1/2 was correlated with decreased levels of 25(OH)D in patients with SLE (r=−0.40, p=0.03; figure 2A). No correlation was seen between B cell activity and 25(OH)D levels in controls (pERK1/2: r=−0.05, p=0.79). Additionally, patients with 25(OH)D levels <20 ng/ml had increased mean (SD) pERK1/2 compared with patients with 25(OH)D levels >20 ng/ml (figure 2B; 56.4 (24.2) vs 39.6 (33.2); p=0.045, unpaired t test with Welch's correction of log-transformed data). Interestingly, there was also a trend for vitamin D-deficient patients to be anti-Ro positive compared with non-deficient patients (59% vs 20%) (OR 5.8, 95% CI 0.99 to 33.8; p=0.061, Fisher exact test). Anti‑Ro-positive patients in this small cohort were not more likely to have documented photosensitivity than anti-Ro-negative patients (60% vs 77%; p=0.450, Fisher exact test). This trend was not seen with any other measured lupus-associated antibody (dsDNA, La, Sm, nRNP, ribo P, aPL).

Lower 25-hydroxyvitamin D (25(OH)D) levels are associated with increased B cell activation in patients with systemic lupus erythematosus (SLE). (A) Increased B cell activation (as measured by phospho-ERK (pERK1/2)) was correlated with decreased 25(OH)D levels in patients with SLE (r=−0.40, p=0.03). (B) Patients with SLE with 25(OH)D levels <20 ng/ml had higher B cell activation (as measured by pERK1/2) than patients with 25(OH)D levels >20 ng/ml; *p=0.045 (unpaired t test with Welch's correction of log-transformed data). Error bars indicate SEM.
Vitamin D-deficient patients with SLE have increased IFNα activity
Serum IFNα activity was measured in serum samples from patients with SLE and compared between patients with 25(OH)D levels >20 ng/ml (n=10) and <20 ng/ml (n=22). Vitamin D-deficient patients had increased IFNα activity compared with patients with 25(OH)D >20 ng/ml (p=0.02, unpaired t test with Welch's correction of log-transformed data; figure 3A). Vitamin D-deficient patients had a mean (SD) serum IFNα activity of 3.5 (6.6) compared with 0.34 (0.33) in non-vitamin D-deficient patients. Anti-Ro, anti-dsDNA and anti-nRNP have each been shown to be independently associated with high serum IFNα activity and, in addition, have been shown to influence serum IFNα activity additively.32 33 We therefore analysed the number of lupus-associated autoantibody specificities present in patients with high and low IFNα activity. Patients with SLE who had high IFNα activity (>1 SD above the mean of healthy controls) had an increased number of lupus-associated autoantibody specificities compared with those who had low IFNα activity (<1 SD above the mean of healthy controls) (2.6 vs 0.9; p=0.002, unpaired t test; figure 3B).
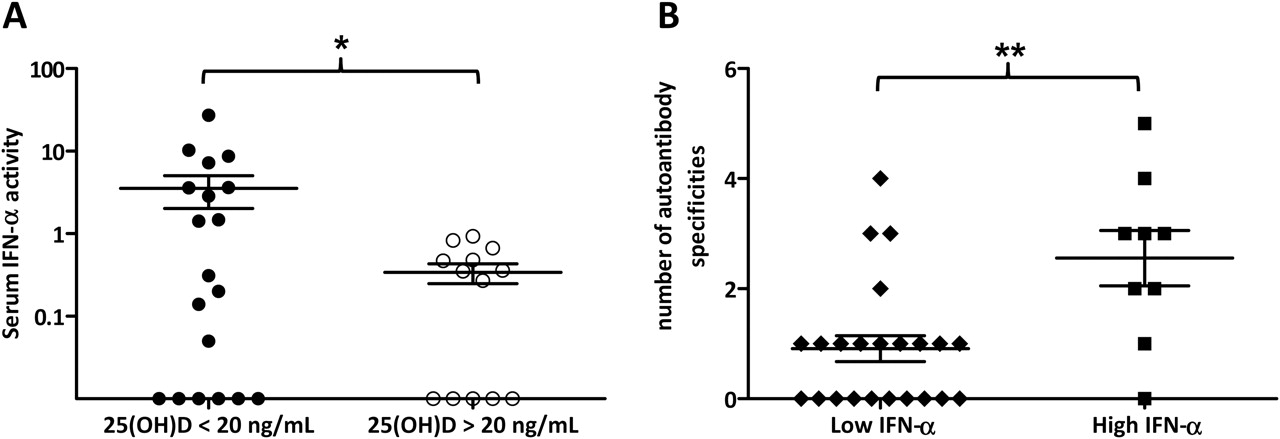
Increased serum interferon α (IFNα) activity is associated with vitamin D deficiency and increased number of autoantibody specificities. (A) Patients with systemic lupus erythematosus (SLE) with 25-hydroxyvitamin D (25(OH)D) <20 ng/ml had mean (SD) serum IFNα activity of 3.53 (6.56) compared with 0.34 (0.33) in patients with 25(OH)D >20 ng/ml. *p=0.02 (unpaired t test with Welch's correction of log-transformed data). (B) Patients with SLE with low IFNα activity (IFNα activity <1 SD above the mean of healthy controls) had fewer mean number of autoantibody specificities than patients with high IFNα activity (IFNα activity >1 SD above the mean of healthy controls): 0.9 vs 2.1. **p=0.002 (unpaired t test). Errors bars indicate SEM.
To determine whether the relationship between 25(OH)D and IFNα activity was dependent on the number of autoantibody specificities, various statistical models were compared using logistic regression. In these models the outcome variable was IFNα activity, with high levels defined as IFNα activity >1 SD above the mean of healthy controls and low levels defined as IFNα activity <1 SD below the mean. The number of lupus-associated autoantibody specificities and 25(OH)D values are the predictor variables. In single-predictor logistic regression models, both 25(OH)D (p=0.033) and number of autoantibody specificities (p=0.023) are significantly associated with IFNα activity. The best fitting single-predictor model was compared with a model with both 25(OH)D and number of autoantibody specificities as predictors using the likelihood ratio test, and the fit was significantly improved with the two-predictor model (χ2=3.931, p=0.047). This suggests that, although the number of autoantibody specificities and 25(OH)D levels are correlated (r2=0.138, p=0.037), both are good independent predictors of IFNα activity.
Discussion
Although increasing evidence supports the role of vitamin D in modulating the immune system, many details remain to be elucidated. While specific mechanisms have been discovered for the influence of vitamin D on innate immunity, the potential role for vitamin D in the adaptive immune system is still not clear. Several mechanisms exist by which vitamin D could potentially modulate adaptive immune responses, including modulation of antigen presentation as well as direct actions on T and B lymphocytes.
This study suggests a role for vitamin D in modulating autoantigenic immune responses. Vitamin D deficiency was associated with an increased presence of autoantibodies in healthy controls. While vitamin D deficiency has been reported in many autoimmune diseases, this is the first observation in ANA-positive healthy individuals. This finding is interesting because patients with autoimmune disease, especially those with SLE, possess many risk factors for vitamin D deficiency whereas healthy controls do not. This provides epidemiological evidence to suggest that vitamin D deficiency in autoimmunity is not solely a consequence of lifestyle changes associated with the disease. A longitudinal study would determine if replenishing vitamin D to sufficient levels would cause a decrease in ANA titres.
We also showed evidence of a relationship between vitamin D levels in patients with SLE and the magnitude of B lymphocyte activation in PBMCs, as determined by pERK1/2 levels. This observation suggests that vitamin D deficiency could be playing a role in the B cell hyperactivity seen in patients with SLE, thus contributing to an increased production of autoantibodies. Interestingly, this same correlation was not seen in the controls, suggesting a potential gene–environment interaction between SLE susceptibility genes and vitamin D. In this manner, vitamin D deficiency would contribute to B cell hyperactivation and autoantibody production in genetically susceptible individuals. Vitamin D deficiency is probably not sufficient to cause B cell hyperactivation and autoantibody production, but rather is a contributing factor along with other genetic and environmental risks.
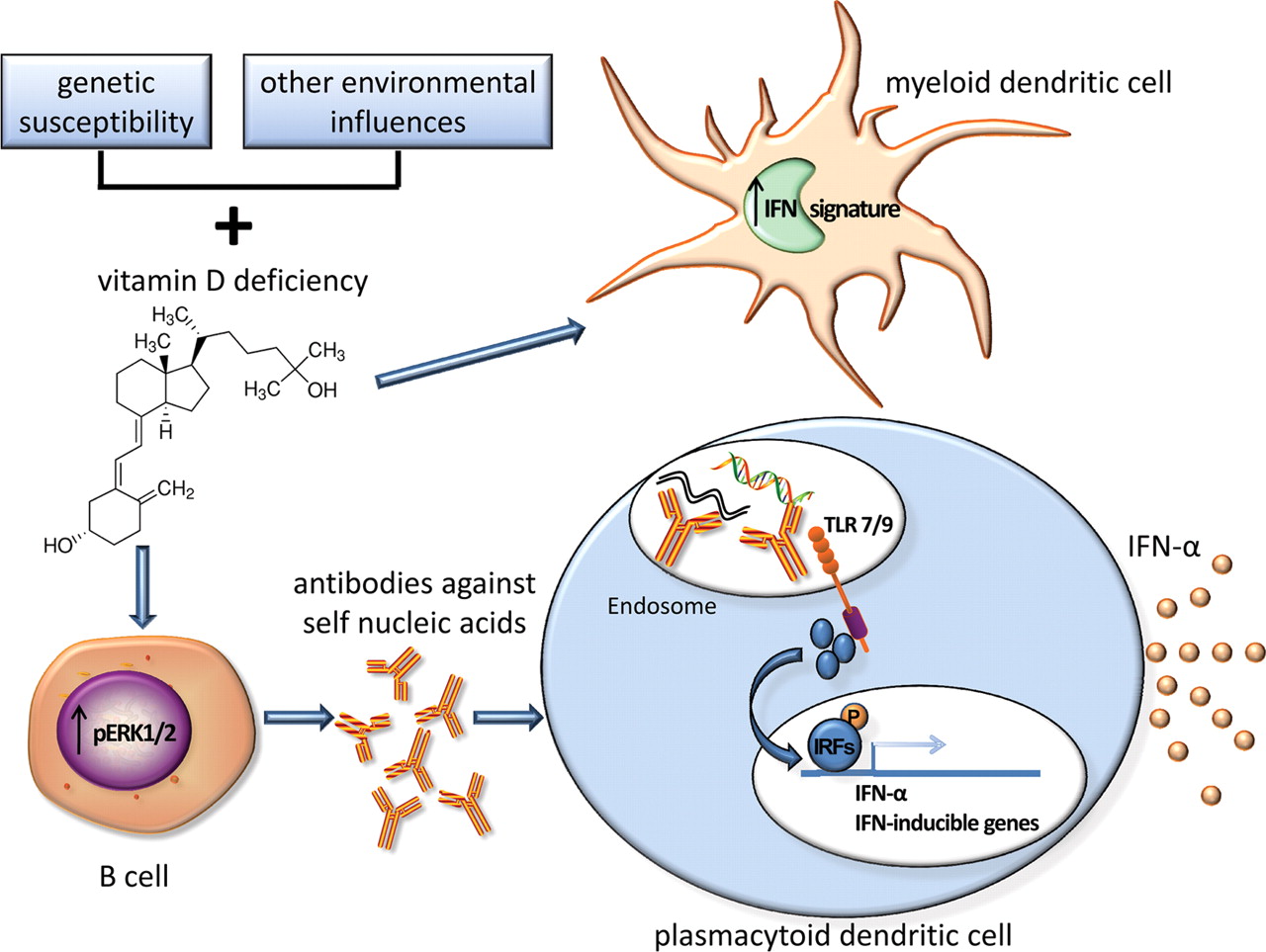
The hypothesis that vitamin D deficiency contributes to increased B cell activation in patients with SLE and increased production of autoantibodies, in particular those directed against nucleic acids, provides a mechanism for the association of vitamin D deficiency with increased IFNα activity (working hypothesis shown in figure 4). Nucleic acids contained within autoantibody immune complexes can activate TLRs, thus promoting IFNα production from plasmacytoid dendritic cells (pDCs) in patients with SLE.34 Vitamin D may bind vitamin D receptors present in pDCs and influence their IFNα production, although it has been shown that 1,25(OH)2D did not modulate IFNα production by pDCs in vitro at the concentrations used.35 Additionally, it has been shown that vitamin D suppresses the expression of the IFN signature in myeloid-derived dendritic cells (MDDCs). This was accomplished using MDDCs from patients with SLE as well as MDDCs generated from controls which had been incubated with plasma from patients with SLE.24 25

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Potential mechanism for the role of vitamin D in B cell hyperactivity, autoantibody production and interferon α (IFNα) activity. Together with genetic susceptibility and other environmental factors, vitamin D deficiency could contribute to increased B cell activation and autoantibody production. This increase in autoantibody production, specifically of antibodies directed against self-nucleic acids, could lead to an increase in IFNα production by plasmacytoid dendritic cells via Toll-like receptor (TLR) signalling mediated by immune complexes. Vitamin D deficiency could also contribute to an increased IFN signature in myeloid dendritic cells.
Experimental studies have also shown that 1,25(OH)2D is able to skew the T cell compartment into a more anti-inflammatory and regulated state, with inhibitory actions on Th1 and Th17 cells and promoting the development of CD4 CD25 Foxp3+ regulatory T cells (Treg).17 Vitamin D has been shown to induce tolerogenic dendritic cells, which promote Treg cell development and enhance recruitment of Treg cells to inflammatory sites.17 Thus, there are several different possible pathways by which vitamin D can influence the pathogenesis of SLE, B cell activity and autoantibody production.
The fact that both ANA-positive healthy individuals and patients with SLE have decreased vitamin D suggests that the mechanism operates early in the steps to SLE development, before the appearance of clinical findings. Beyond considering the molecular mechanisms by which vitamin D deficiency would predispose to autoimmunity, the extraordinarily high prevalence of vitamin D deficiency in ANA-positive healthy individuals and patients with SLE strongly suggests that repletion with vitamin D should be considered. A recent study sought to examine the effects of oral vitamin D supplementation on disease activity in SLE; however, more than 70% of the patients in the study still had insufficient levels of vitamin D after 2 years of treatment.36 While this study did not report any improvements in SLE disease activity after oral administration of vitamin D, interpretation of the results is limited by the small percentage of patients with SLE who achieved adequate 25(OH)D levels. Although no definitive study has been published demonstrating a beneficial effect of vitamin D supplementation on SLE disease severity, current knowledge supports vitamin D replacement for calcium homeostasis, bone health and potential immune system benefits.
Acknowledgments
The authors would like to thank all the study participants for their time and commitment to the study as well as the referring physicians: Drs C Carson, A A Kumar, L Zacharias and physician assistants T Aberle and J K Shoemaker. The authors would also like to thank J Anderson, W Klein, G Vidal and J Levin for their technical assistance, as well as S Stewart for his assistance with the statistical analysis.
References
Supplementary materials
Web Only Data
Files in this Data Supplement:
Footnotes
-
Funding This work was supported by the National Institutes of Health (NIAID: HHSN266200500026C, AR058554, RR015577, AI082714, AI24717, AR24260, AI083194, AR052364 and AR053483), Kirkland Scholar Award Program at The Hospital for Special Surgery in New York City which is funded exclusively by Rheuminations, Inc., a non-profit foundation dedicated to supporting research leading to the treatment and cure of lupus, OMRF J Donald Capra Fellowship Support, US Department of Veteran Affairs and the OMRF Lou C Kerr Chair in Biomedical Research.
-
Competing interests JBH has served as a consultant for BioRad and owns stock in IVAX Diagnostics. All other authors have no competing interest.
-
Ethics approval This study was conducted with the approval of the Oklahoma Medical Research Foundation (OMRF) and the University of Oklahoma Health Sciences Center.
-
Provenance and peer review Not commissioned; externally peer reviewed.