Article Text

Abstract
Objectives Myositis is a heterogeneous family of diseases that includes dermatomyositis (DM), antisynthetase syndrome (AS), immune-mediated necrotising myopathy (IMNM), inclusion body myositis (IBM), polymyositis and overlap myositis. Additional subtypes of myositis can be defined by the presence of myositis-specific autoantibodies (MSAs). The purpose of this study was to define unique gene expression profiles in muscle biopsies from patients with MSA-positive DM, AS and IMNM as well as IBM.
Methods RNA-seq was performed on muscle biopsies from 119 myositis patients with IBM or defined MSAs and 20 controls. Machine learning algorithms were trained on transcriptomic data and recursive feature elimination was used to determine which genes were most useful for classifying muscle biopsies into each type and MSA-defined subtype of myositis.
Results The support vector machine learning algorithm classified the muscle biopsies with >90% accuracy. Recursive feature elimination identified genes that are most useful to the machine learning algorithm and that are only overexpressed in one type of myositis. For example, CAMK1G (calcium/calmodulin-dependent protein kinase IG), EGR4 (early growth response protein 4) and CXCL8 (interleukin 8) are highly expressed in AS but not in DM or other types of myositis. Using the same computational approach, we also identified genes that are uniquely overexpressed in different MSA-defined subtypes. These included apolipoprotein A4 (APOA4), which is only expressed in anti-3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) myopathy, and MADCAM1 (mucosal vascular addressin cell adhesion molecule 1), which is only expressed in anti-Mi2-positive DM.
Conclusions Unique gene expression profiles in muscle biopsies from patients with MSA-defined subtypes of myositis and IBM suggest that different pathological mechanisms underly muscle damage in each of these diseases.
- dermatomyositis
- polymyositis
- autoantibodies
- autoimmune diseases
- autoimmunity
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Different types of myositis are likely to have unique pathological mechanisms.
What does this study add?
Machine learning algorithms can be trained on transcriptomic data to classify muscle biopsies from patients with dermatomyositis, antisynthetase syndrome, immune-mediated necrotising myopathy and inclusion body myositis.
Recursive feature elimination can be used to determine which genes are most important for the machine learning algorithms to classify the muscle biopsies.
Only antisynthetase syndrome muscle biopsies express high levels of CAMK1G (calcium/calmodulin-dependent proteinkinase IG), EGR4 (early growth response protein 4) and CXCL8 (interleukin 8).
APOA4 (apolipoproteinA4), a gene involved in cholesterol metabolism, is uniquely overexpressed in anti-HMGCR myopathy, which can be triggered by statins.
MADCAM1 (mucosalvascular addressin cell adhesion molecule 1), which recruits lymphocytes to target tissues, is uniquely overexpressed in muscle biopsies from those with anti-Mi2-positive dermatomyositis.
How might this impact on clinical practice or future developments?
Gene expression profiling of muscle biopsies from individual myositis patients may identify specific pathological pathways that could be used to tailor therapies.
Introduction
The idiopathic inflammatory myopathies (IIM) are a heterogeneous family of diseases that includes six major types: dermatomyositis (DM), antisynthetase syndrome (AS), immune-mediated necrotising myopathy (IMNM), inclusion body myositis (IBM), polymyositis and overlap myositis.1 Furthermore, 50% to 80% of IIM patients have myositis-specific autoantibodies (MSAs) that define phenotypically distinct IIM subtypes.2 3
Muscle biopsies from patients with each major type of myositis have distinctive pathological features. For example, perifascicular myofibre atrophy and/or necrosis is a characteristic feature of both DM and AS, IMNM biopsies have abundant scattered necrotic myofibres and IBM muscle biopsies usually include myofibres with cytoplasmic vacuoles.4 However, histological features that can reliably distinguish between DM and AS have not been identified. Similarly, histological features cannot reliably be used to distinguish between different MSA-defined subtypes of DM or IMNM. Thus, it remains unclear whether different pathological pathways lead to muscle damage in the different myositis types and MSA-defined subtypes.
The advent of gene chip microarray and next-generation sequencing technologies has facilitated the use of myositis muscle biopsy gene expression profiles to identify pathological pathways. For example, microarray analysis led to the discoveries that type I and type II interferon (IFN)-inducible genes are upregulated in muscle biopsies from patients with DM5 and IBM,6 7 respectively. However, disease-specific gene expression profiles have not been fully described in patients with IMNM, AS or any of the autoantibody-defined subtypes of DM. Furthermore, little attention has been given to genes that are differentially expressed between patients with different types and subtypes of myositis.5 8–10 In the current study, we trained machine learning algorithms to classify muscle biopsies using transcriptomic data from normal, IBM and MSA-positive muscle biopsies; biopsies from 20% to 50% of myositis patients who are MSA-negative were not included in this study. We then used recursive feature elimination to identify novel disease-specific gene expression patterns that may be pathologically relevant in DM, AS, IMNM, IBM and MSA-defined subtypes of myositis.
Materials and methods
Patients, samples and autoantibody testing
Muscle biopsies obtained from subjects enrolled in institutional review board (IRB)-approved longitudinal cohorts from the National Institutes of Health (IRB number 91-AR-0196), the Johns Hopkins Myositis Center (IRB number NA_00007454), the Clinic Hospital (Barcelona; IRB number HCB/2015/0479) and the Vall d’Hebron Hospital (Barcelona; IRB number PR (AG) 68/2008) were included in the study if the patients fulfilled IBM criteria according to Lloyd,11 or had one of the following MSAs: anti-NXP2, anti-Mi2, anti-TIF1γ, anti-MDA5, anti-HMGCR, anti-SRP or anti-Jo1. Autoantibody testing was performed as previously described for anti-HMGCR and by line blot for the others (EUROLINE Myositis Profile 4). Patients were classified as having the AS if they had autoantibodies against Jo-1 and fulfilled Connors’ AS criteria,12 in the DM group if they had autoantibodies recognising Mi2, NXP2, TIF1γ or MDA5 and in the IMNM group if they tested positive for anti-SRP or anti-HMGCR autoantibodies. Normal muscle biopsies were obtained from the Johns Hopkins Neuromuscular Pathology Laboratory (n=10) and the Skeletal Muscle Biobank of the University of Kentucky (n=10).
Patient consents
Written informed consent was obtained from each participant.
Human muscle biopsy processing, human skeletal muscle cell culture and mouse muscle injury
See online supplementary methods.
Supplemental material
RNA-sequencing
RNA-sequencing (RNA-seq) was performed as previously described.13 Briefly, RNA was prepared using TRIzol. Libraries were prepared using the NeoPrep system according to the TruSeq Stranded mRNA Library Prep protocol (Illumina) and sequenced using the Illumina HiSeq 2500 or 3000. Reads were aligned using the STAR V.2.5 25, the abundance of each gene was quantified using StringTie V.1.3.3.26 and the differential gene expression was performed using DESeq2 V.1.20 (online supplementary methods). The Benjamini-Hochberg correction was used to adjust for multiple comparisons and a corrected p value (q-value) of 0.05 or less was considered statistically significant.
Pathway analysis
We used Ingenuity Pathway Analysis V.01–07 and genes with a q-value below 0.05 and an expression ratio greater than 2 in each group compared with the rest of the biopsies were included in the analysis. Immunologic pathways with a z-score over 2 were selected.
RNA-seq based classification
To find the ability of RNA-seq data to classify different types of myositis we first tested several classification models. Next, we performed stratified cross-validation to estimate the accuracy of each model. All steps were performed using Python V.3.6.3. NumPy V.1.13.3 and pandas V.0.20.3 were used for data wrangling and basic statistical calculations, respectively (online supplementary methods).
Those genes with significantly differential expression levels in one group compared with the rest of the biopsies were included in each model. The sample was split into a training set containing two out of three of the observations and a test set containing the remaining one out of three. The training set was used to build the classificatory models and the testing set to evaluate the accuracy of the model. The machine learning models were developed using the package scikit-learn V.0.19.1. Models were built using two out of three random resamples of the data and tested in the remaining one out of three. The accuracy of classifying each of the myositis subsets was determined based on the mean and 95% CI of 1000 resampling cycles (online supplementary methods).
Recursive feature elimination was applied to the whole data set to rank each gene according to how useful it was for the model to differentiate the different patient groups. The recursive feature elimination technique was applied through its implementation in scikit-learn V.0.19.1 (online supplementary methods).
Statement of patient and public involvement
Neither patients nor the public were involved in the design, conduct, reporting or dissemination of this research.
Data availability statement
De-identified RNA-seq data will be made available on request to Dr Andrew Mammen (andrew.mammen@nih.gov).
Results
Machine learning models accurately classify muscle biopsies
Muscle biopsy specimens were available from 119 myositis patients including 39 with DM (11 anti-Mi2-positive, 12 anti-NXP2-positive, 11 anti-TIF1γ-positive and 5 anti-MDA5-positive), 49 with IMNM (9 anti-SRP-positive and 40 anti-HMGCR-positive), 18 with anti-Jo1-positive AS and 13 with IBM. Twenty normal muscle biopsy specimens were used as comparators. Expression levels of all genes were determined for each sample by RNA-seq. Details regarding the patients and their muscle biopsy features are found in online supplementary table 1. Expression levels of genes associated with immune cells, regenerating myofibres and mature skeletal muscle are found in online supplementary figure 1.
Supplemental material
Supplemental material
First, we identified those genes with statistically significant differential expression in controls and each major type of myositis compared with the rest of the groups. A total of 10 141 differentially expressed genes were identified and the top 10 for each group are listed in table 1. For example, the interferon-inducible gene ISG15 is the top differentially expressed gene in both DM and normal muscle biopsies; it is expressed at 43-fold higher levels in DM compared with the rest of the groups and at 17-fold lower levels in normal biopsies compared with the rest of the groups.
Genes differentially expressed in muscle biopsies from each major type of myositis and controls compared with the rest of the samples
Supplemental material
To determine whether machine learning programmes could use transcriptomic data to accurately classify patients into each major type of myositis or the control group, all differentially expressed genes were included in each of 10 machine learning models (online supplementary methods). From among the models tested, the linear support vector machine (SVM) model performed the best with accuracies of 91% or greater to identify normal DM, AS, IMNM and IBM muscle biopsies. (table 2).
A comparison of machine learning models to classify muscle biopsies based on gene expression data. Accuracy and 95% CI in the 1000 test sets of the different machine learning models to classify muscle biopsies into normal muscle tissue (NT), dermatomyositis (DM), antisynthetase syndrome (AS), inclusion body myositis (IBM) or immune-mediated necrotising myopathy (IMNM).
Identifying genes with unique expression patterns in DM, AS, IMNM and IBM
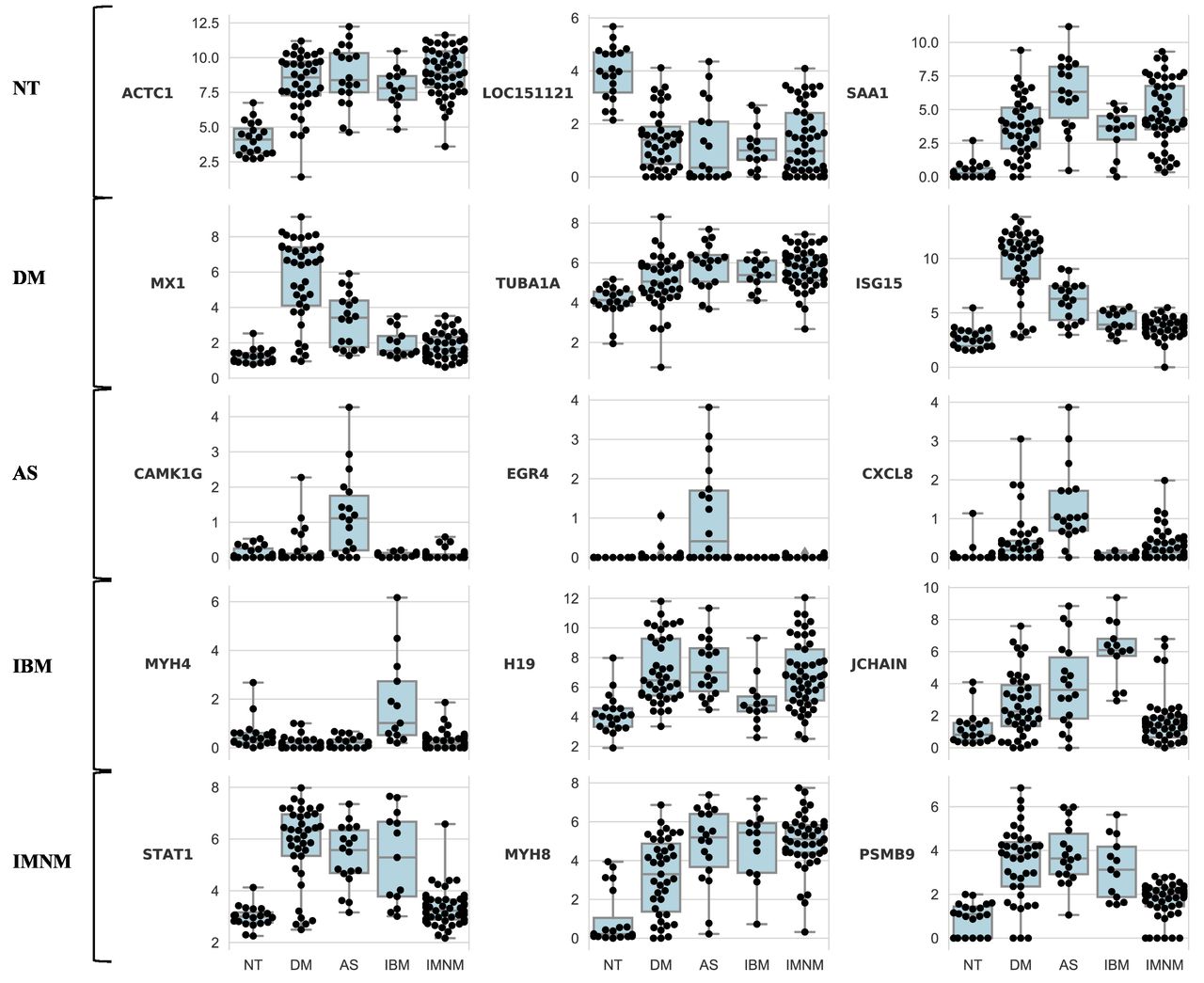
We expected that for each major type of myositis, those genes contributing most to the accuracy of the machine learning classification model would be involved in disease-specific pathological processes. To identify which among the thousands of differentially expressed genes used by the linear SVM model are most useful to classify a biopsy into each type of myositis, we used the recursive feature elimination technique.14 This method systematically eliminates genes with the weakest role in the model, leaving those that are most important to classify muscle biopsies into the correct group. Table 3 lists the 10 genes whose expression levels have the greatest utility to identify samples as belonging to each type of myositis or control group. Figure 1 shows the expression levels of the three most important genes from each group.
The top 10 most useful genes to differentiate biopsy samples using the recursive feature elimination technique on the support vector machine model

Expression levels of those genes most helpful to classify muscle biopsies into each type of myositis. The expression levels of the top three genes used by the support vector machine model to classify muscle biopsies from normal tissue (NT), dermatomyositis (DM), immune-mediated necrotising myositis (IMNM), antisynthetase syndrome (AS) or inclusion body myositis (IBM).
We first sought to validate this approach by determining whether it would identify key genes already known to play roles in DM pathogenesis. As genes upregulated by type I IFN are known to be expressed at high levels in DM muscle,5 15 we expected that expression levels of type I IFN-inducible genes should be important for the linear SVM model. Indeed, high expression levels of type 1 IFN-inducible genes MX1 and ISG15 were among the three most important features used to identify DM muscle biopsies (table 3).
When applied to the AS group, recursive feature elimination identified CAMK1G (calcium/calmodulin-dependent protein kinase IG), EGR4 (early growth response protein 4) and CXCL8 (interleukin 8) as the three most important genes (table 3). Each of these genes is expressed at markedly higher levels in AS than in the other groups (figure 1).
High expression levels of MYH4 (myosin heavy chain 4) and JCHAIN (the joining chain of multimeric IgA and IgM) were among the three most important features used by the linear SVM model to identify samples as belonging to the IBM group (table 3 and figure 1). In addition, the low expression level of H19 (a non-coding RNA) in IBM compared with DM, AS and IMNM (figure 1) appeared to be important for IBM classification.
Expression levels of STAT1 (signal inducer and activator of transcription 1), MYH8 (myosin heavy chain 8) and PSMB9 (proteasome subunit beta 9) were the top features used to classify a muscle biopsy as IMNM (table 3). Based on the patterns of expression (figure 1), the model seems to rely both on the low expression of IFN-inducible genes STAT1 and PSMB9 (expressed at high levels in DM, AS and IBM) as well as the high expression of MYH8 (expressed at low levels in normal muscle) to classify biopsies as IMNM.
The expression levels of ACTC1 (actin alpha cardiac muscle 1), LOC151121 (a non-coding gene) and SAA1 (serum amyloid A1) were the top features used to classify normal muscle biopsies (table 3). Interestingly, normal muscle biopsies were characterised by low levels of ACTC1, which encodes a structural protein expressed during muscle regeneration16 (figure 1). Similarly, the SAA1 gene, which encodes the acute phase reactant serum amyloid A1, was expressed at low levels in normal muscles and high levels in all of the myositis groups. In contrast, LOC151121 was expressed at high levels in normal muscle but at low levels in all the myositis groups (figure 1).
Identifying genes with unique expression patterns in the different subtypes of IMNM and DM
Using the same methodology, we next identified those genes that were most useful to classify biopsies according to the different autoantibody-defined subtypes within IMNM and DM. This revealed that APOA4 (apolipoprotein A4) was selectively expressed in IMNM patients with anti-HMGCR autoantibodies (figure 2). Similarly, MADCAM1 (mucosal vascular addressin cell adhesion molecule 1) was exclusively detectable in DM patients with anti-Mi2 autoantibodies (figure 2).

Genes selectively upregulated in different autoantibody-defined subtypes of myositis. APOA4 and MADCAM1 are selectively overexpressed (log2(FPKM+1)) in anti-HMGCR IMNM (q-value compared with SRP: 0.0009) and anti-Mi2 DM (q-value compared with other DM antibodies: 2.9E-9), respectively.APOA4,apolipoprotein A4; AS, antisynthetase syndrome; DM, dermatomyositis; HMGCR, anti-HMGCR IMNM; IBM, inclusion body myositis; IMNM, immune-mediated necrotising myopathy; Jo1, anti-Jo1AS; MADCAM1, mucosal vascular addressin cell adhesion molecule 1; MDA5, anti-MDA5DM; Mi2, anti-Mi2 DM; NT,normal muscle tissue; NXP2; anti-NXP2DM; SRP, anti-SRP IMNM; TIF1, anti-TIF1gDM.
Pathway analysis
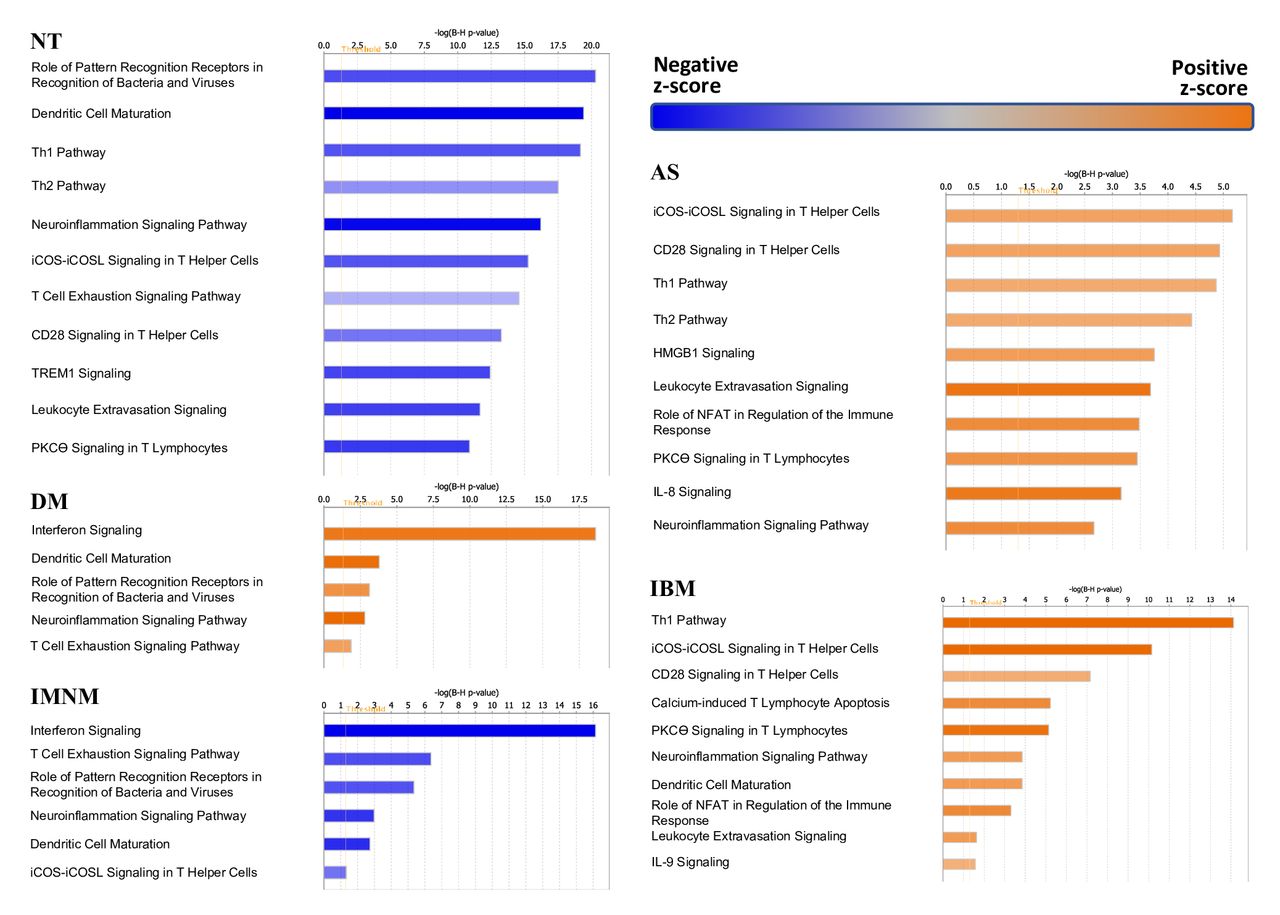
To gain further insight into the biological processes that distinguish each group compared with the others, we performed pathway analyses. For each analysis, we included the set of genes differentially expressed by at least twofold in the type of myositis (or control) compared with the rest of the biopsies. Pathways annotated as related to the ‘cellular immune response’, ‘cytokine signalling and ‘humoral immune response’ (ie, immunological pathways) were included in each analysis.
As expected, ‘interferon signalling was the top over-represented immunologic pathway in DM (figure 3). The AS and IBM biopsies shared the same top three over-represented pathways that were not included DM, IMNM or control biopsies. These included the T cell pathways ‘ICOS-ICOSL signalling in T helper cells’, ‘CD28 signalling in T helper cells’ and the ‘Th1 pathway’. No immunological pathways were over-represented in IMNM biopsies. Rather, IMNM biopsies, like control biopsies, were notable for the under-representation of pathways that were important in DM, AS and/or IBM.

{kind=link}
{kind=link}
{kind=link}
Pathway analysis in myositis and normal muscle biopsies. The top 10 pathways of the different muscle biopsy groups are shown.AS,antisynthetase syndrome; DM, dermatomyositis; IMNM, immune-mediated necrotisingmyopathy; IBM, inclusion body myositis; NT, normal tissue.
Muscle regeneration genes are among the top differentially expressed genes in IMNM and are also overexpressed in other types of myositis
To classify biopsies as IMNM, linear SVM relied on the relative underexpression of genes expressed at high levels in other forms of myositis (eg, STAT1 and PSMB8)15 rather than on genes that were uniquely overexpressed in IMNM. To further investigate pathological processes important for IMNM, we considered the known functions of the top 10 overexpressed genes in biopsies from these patients (table 4). Interestingly, several of these are known to play a role in skeletal muscle differentiation and/or muscle repair. For example, ACTC1 encodes alpha-actin which is expressed in early adult skeletal muscle.16 Similarly, tenascin C (TNC) encodes an extracellular matrix protein that is expressed in actively remodelling musculoskeletal tissue.17
The top 10 upregulated genes in each type of myositis compared with normal biopsies
To determine whether the other most overexpressed genes in IMNM play a role in muscle regeneration, we analysed their expression levels in cultured human myoblasts as they differentiated to form myotubes. Each gene was expressed at low levels in myoblasts and at high levels in differentiating myotubes (online supplementary figure 2). Similarly, these genes were expressed at low levels in healthy mouse muscle, but at high levels in regenerating mouse muscles following a muscle injury (online supplementary figure 3). This pattern suggests that these genes are expressed as part of the muscle regeneration process induced by necrosis in IMNM muscle. Since regeneration is also a common feature of muscle biopsies from those with DM, AS and IBM, we expected that muscle biopsies from each of these types of myositis should also have high levels of the genes overexpressed in IMNM. Indeed, even though they were not among the top 10 overexpressed genes in the other groups, each of these genes was highly expressed in the other types of myositis muscle but not control muscle (online supplementary figure 4).
Supplemental material
Supplemental material
Supplemental material
We next considered the known functions of the top 10 upregulated genes in DM, AS and IBM compared with control muscle (table 4). Consistent with prior studies, many of the top 10 differentially expressed genes in muscle biopsies from DM patients are inducible by interferon type I (eg, ISG15,18 19 IFI620 and MX121) (table 4). Similarly, several of the most overexpressed genes in AS and IBM muscle biopsies are interferon type II inducible genes (eg, PSMB8,22 GBP2 and GBP123 24) (table 4).
Discussion
In this study, we showed that machine learning algorithms trained on transcriptomics data could accurately classify myositis muscle biopsies from IBM and antibody-positive DM, AS and IMNM patients. This demonstrates that these IIM types have unique gene expression profiles. Indeed, by applying recursive feature elimination to the machine learning algorithms we identified novel gene markers (eg, CAMK1G, EGR and CXCL8) that are uniquely expressed in AS but not DM, even though these two diseases can be histologically indistinguishable. Moreover, we also identified genes (eg, ACTC1 and SSA1) that are overexpressed in all types of myositis studied here but not in normal muscle. Finally, we confirmed previous observations related to the pathogenesis of myositis, including the role of interferon pathways in DM,5 15 the prominence of muscle regeneration in IMNM25 and the presence of plasma cells in IBM (as evidenced by overexpression of JCHAIN, a plasma cell marker).26 27
We applied the same computational approach to identify genes that are uniquely upregulated in patients with different MSA-defined IIM subtypes. For example, although anti-SRP and anti-HMGCR myopathy muscle biopsies are histologically identical, we identified the APOA4 gene as being exclusively upregulated in the latter subtype of IMNM. Since statin exposure is a risk factor for developing anti-HMGCR myopathy but not other types of myositis,28 it is of interest that APOA4, which contributes to reverse cholesterol transport by facilitating the movement of cholesterol from the periphery to the liver for excretion,29 is only upregulated in anti-HMGCR myopathy muscle biopsies.
We also found that different MSA-defined DM subtypes had different gene expression profiles. For example, MADCAM1 was uniquely expressed in muscle biopsies from DM patients with anti-Mi2 autoantibodies. Of note, MADCAM1 is expressed on endothelial surfaces in the intestine where it mediates the migration of lymphocytes into the gut by binding to α4β7 integrin found on the surface of CD4+ and CD8+ T cells.30 Since MADCAM1 recruits inflammatory cells to the gut in patients with colitis, we hypothesise that it could play a similar role in anti-Mi2-positive DM patients, who have more lymphocytic invasion of muscle fibres than DM patients with other autoantibodies.31 This could have therapeutic implications since drugs that target the MADCAM1/α4β7 pathway have already been developed.
This study was not designed to directly compare the performance of machine learning algorithms using muscle biopsy transcriptomic data with the analysis of histological features to diagnose different types of myositis. Still, the current study suggests that machine learning algorithms would fare favourably in such a comparison. For example, only 72% of biopsies from the included DM patients had perifascicular atrophy,31 the key feature required for histological diagnosis of DM.32 Nonetheless, the SVM algorithm diagnosed DM based on the muscle biopsy transcriptome with an accuracy of 92%. This raises the possibility that, with the availability of gene expression profile data collected from a large number of patients with different types of myopathy, machine learning algorithms could be diagnostically useful.
This study was limited in that we did not include muscle biopsies from all types of myositis. Indeed, we excluded biopsies from patients with polymyositis, overlap myositis and MSA-negative forms of myositis. Furthermore, our analysis was restricted to gene expression data and did not include analyses of the corresponding proteins. Nonetheless, by applying machine learning algorithms to muscle biopsy transcriptomic data, we have demonstrated that DM, AS, IMNM and IBM can be distinguished based on their unique gene expression patterns. Furthermore, by applying recursive feature elimination to these classification models, we not only confirmed known pathological pathways in IIM, such as the role of type I interferon in DM, we also identified novel genes that are uniquely upregulated in other types and MSA-defined subtypes of myositis. We expect this computational approach could be useful for analysing transcriptomic data from other autoimmune conditions in which there are different types and subtypes of the disease.
Supplemental material
Acknowledgments
The authors thank Dr Gustavo Gutierrez-Cruz, Dr Stefania Dell’Orso and Faiza Naz from the NIAMS sequencing facility for all their technical collaboration in making the RNA-seq libraries and sequencing them, and the University of Kentucky Center for Muscle Biology for providing normal human muscle samples for the study.
References
Footnotes
Handling editor Josef S Smolen
Contributors All authors have met these four criteria: Substantial contributions to the conception or design of the work, or the acquisition, analysis or interpretation of data. Drafting the work or revising it critically for important intellectual content. Final approval of the version published. Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding This research was supported in part by the Intramural Research Programme of the National Institute of Arthritis and Musculoskeletal and Skin Diseases and the National Institute of Environmental Health Sciences of the National Institutes of Health. The Myositis Research Database and Dr LC-S are supported by the Huayi and Siuling Zhang Discovery Fund. IPF's research was supported by a Fellowship from the Myositis Association. The authors also thank Dr Peter Buck for support.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval This study was approved by the Institutional Review Boards at participating institutions.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request. De-identified RNA-seq data will be made available upon request to Dr Andrew Mammen at andrew.mammen@nih.gov.