Article Text

Abstract
Objectives To assess the efficacy and safety of abatacept, a selective T-cell costimulation modulator, in a phase III study in psoriatic arthritis (PsA).
Methods This study randomised patients (1:1) with active PsA (~60% with prior exposure to a tumour necrosis factor inhibitor) to blinded weekly subcutaneous abatacept 125 mg (n=213) or placebo (n=211) for 24 weeks, followed by open-label subcutaneous abatacept. Patients without ≥20% improvement in joint counts at week 16 were switched to open-label abatacept. The primary end point was the proportion of patients with ≥20% improvement in the American College of Rheumatology (ACR20) criteria at week 24.
Results Abatacept significantly increased ACR20 response versus placebo at week 24 (39.4% vs 22.3%; p<0.001). Although abatacept numerically increased Health Assessment Questionnaire–Disability Index response rates (reduction from baseline ≥0.35) at week 24, this was not statistically significant (31.0% vs 23.7%; p=0.097). The benefits of abatacept were seen in ACR20 responses regardless of tumour necrosis factor inhibitor exposure and in other musculoskeletal manifestations, but significance could not be attributed due to ranking below Health Assessment Questionnaire–Disability Index response in hierarchical testing. However, the benefit on psoriasis lesions was modest. Efficacy was maintained or improved up to week 52. Abatacept was well tolerated with no new safety signals.
Conclusions Abatacept treatment of PsA in this phase III study achieved its primary end point, ACR20 response, showed beneficial trends overall in musculoskeletal manifestations and was well tolerated. There was only a modest impact on psoriasis lesions.
Trial registration number ClinicalTrials.gov number, NCT01860976 (funded by Bristol-Myers Squibb).
- Psoriatic arthritis
- DMARDs (biologic)
- T cells
- Treatment
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Psoriatic arthritis (PsA) is an inflammatory arthritis that occurs in up to one-third of patients with psoriasis and is usually diagnosed years after the appearance of psoriatic skin disease.1 2 Although current treatments for PsA benefit many patients, a substantial proportion do not achieve significant improvement in their disease.3–5 Consequently, there remains an unmet need for effective and well-tolerated treatments.
PsA is associated with specific major histocompatibility complex class I molecules that are involved in antigen presentation to T cells, which are implicated in disease pathogenesis.6 Abatacept, a selective T-cell costimulation modulator, is a soluble fusion protein comprising the extracellular domain of human cytotoxic T-lymphocyte-associated antigen-4 linked to the modified Fc (hinge, CH2 and CH3 domains) portion of human immunoglobulin G1.7 8 By selectively modulating the CD28 costimulatory signal required for full T-cell activation, abatacept blocks the process that triggers the inflammatory cascade and, therefore, is a potential therapy for PsA with a distinct mechanism of action upstream of currently available agents.4 7 Abatacept is an approved treatment for rheumatoid arthritis (RA) and juvenile idiopathic arthritis, with an established acceptable safety profile.9–14
Data have previously been reported from a phase II, dose-ranging study of abatacept in patients with active PsA and prior exposure to disease-modifying antirheumatic drugs (DMARDs), 37% of whom had previously received tumour necrosis factor inhibitor (TNFi). At 6 months, the dose of 10 mg/kg given intravenously every 4 weeks showed the greatest increase in the proportion of patients with ≥20% improvement in the American College of Rheumatology criteria (ACR20) versus placebo (48% vs 19%, respectively; p=0.006).15 The proportion of patients achieving a Health Assessment Questionnaire–Disability Index (HAQ-DI) response (reduction from baseline score ≥0.3) at 6 months was also increased in the 10 mg/kg group versus placebo (45% vs 19%, respectively). In addition, trends towards improvements over placebo were seen in joint damage, based on MRI. Following these results, the phase III Active PSoriaTic Arthritis RAndomizEd TriAl (ASTRAEA) Study was conducted to assess the efficacy and safety of abatacept in patients with active PsA, using a more convenient subcutaneous 125 mg weekly dose that has shown therapeutic equivalence to intravenous dosing with 10 mg/kg every 4 weeks in RA.16
Methods
Study design and oversight
This ongoing phase III study (total study duration including long-term extension, 729 days) was initiated in June 2013 and conducted across 76 centres worldwide (ClinicalTrials.gov number, NCT01860976). Clinical and radiographic database locks were in August and October 2015 (24-week analysis) and in March and April 2016 (1-year analysis), respectively. Using a central interactive voice response system, patients were randomly assigned (1:1) in a double-blind manner to receive subcutaneous abatacept 125 mg weekly or matched placebo for 24 weeks. Randomisation was stratified globally (rather than at site level) by factors that were considered to potentially impact results, including current methotrexate use, prior TNFi use and whether plaque psoriasis involved ≥3% of body surface area (BSA). Within each stratum, permuted block randomisation was conducted with a block size of two. Patients who had not achieved ≥20% improvement in swollen and tender joint counts from baseline to week 16 were switched to open-label abatacept weekly (early escape (EE)) for 28 weeks (total study time for these patients, 44 weeks). At week 24, all remaining patients transitioned to the open-label period and received subcutaneous abatacept weekly for 28 weeks (total study time, 52 weeks). At the end of the open-label period, patients had the option of entering a 1-year, long-term extension.
The study was conducted in accordance with the Declaration of Helsinki, International Conference on Harmonisation Guidelines for Good Clinical Practice and local regulations. An Institutional Review Board or Independent Ethics Committee approved the protocol, consent form and any other written information provided to patients. Patients were evaluated by the investigators, and the data were collected and analysed by Bristol-Myers Squibb under the direction of the investigators.
Patients
Enrolled patients were aged ≥18 years, met the classification criteria for PsA17 and had active arthritis (≥3 swollen and ≥3 tender joints), active plaque psoriasis with ≥1 qualifying target lesion ≥2 cm in diameter and inadequate response or intolerance to ≥1 non-biologic DMARD. Concomitant treatment with methotrexate, leflunomide, sulfasalazine or hydroxychloroquine, non-steroidal anti-inflammatory drugs and oral corticosteroids (<10 mg/day) and use of low-potency topical corticosteroids in sensitive areas were permitted (as detailed in online section 1 in the supplementary appendix). To reflect a typical patient population in clinical practice and that of the phase II study,15 both TNFi-naïve and TNFi-exposed patients were enrolled. All patients gave written informed consent prior to study entry.
Supplemental material
Assessments
Arthritis was assessed in 66 joints for swelling and 68 joints for tenderness by ACR response criteria for per cent improvement from baseline18 and post hoc by the Disease Activity Score (DAS)28 (C-reactive protein (CRP)).19 Enthesitis at six locations was evaluated using the Leeds Enthesitis Index (range 0–6)20 and dactylitis by the number of tender and swollen digits with a circumference ≥10% greater than the contralateral digit according to the Leeds Dactylitis Index basic score.21 Physical function was measured using the HAQ-DI (range 0–3).22 Among patients with plaque psoriasis involving ≥3% BSA at baseline, skin lesions were assessed using the Psoriasis Area and Severity Index (PASI; range 0–72).23 PsA disease activity was assessed using the minimal disease activity (MDA) criteria,24 the modified Composite Psoriatic Disease Activity Index,25 the Psoriatic Arthritis Disease Activity Score26 and post hoc for the Disease Activity index for Psoriatic Arthritis (DAPSA).27 Quality of life was evaluated using the Short Form-36 (SF-36)28 and the Dermatology Life Quality Index.29
Plain radiographs of hands and feet were taken at baseline and at weeks 24 and 52 (or weeks 16, 24 and 44 for EE patients). Radiographs were scored independently by two central, trained assessors (and an adjudicator in predefined cases) with experience using the PsA-modified Sharp–van der Heijde (SHS) scoring method (total score 0–528).30 Assessors were blinded to patient identity, treatment, clinical data and order of radiographs. Initially, baseline and week 24 (week 16 for EE) radiographs were scored; in a second round, all radiographs, including week 52 (week 44 for EE), were scored. For joint erosion, joint space narrowing and total score, and the proportion of non-progressors, the mean of the scores from two assessors was used. If one score was missing, then the available score was used. If required, an adjudicator reviewed the images, and the mean of the adjudicator’s total score and the other total score that was closer to the adjudicator’s score was used (>0=progressors, ≤0=non-progressors). Safety was evaluated throughout the study by monitoring of adverse events (AEs) and routine laboratory tests.
Efficacy end points
The primary end point was the proportion of patients with ACR20 responses at week 24. Key secondary end points at week 24, in hierarchical order, were the proportions of patients with an HAQ-DI response (reduction from baseline, ≥0.35), an ACR20 response in the TNFi-naïve and TNFi-exposed subgroups and a radiographic non-progression (change from baseline score, ≤0) according to PsA-modified total SHS score. Other secondary end points at week 24 included the proportion of patients with ≥50% and ≥70% improvement in ACR criteria (ACR50 and ACR70, respectively), the proportion who achieved ≥50% improvement in PASI score from baseline (PASI 50) and the mean change from baseline in SF-36 physical and mental component summary scores. Prespecified exploratory end points and post hoc analyses are described in online section 2 in the supplementary appendix.
Supplemental material
Statistical analysis
A hierarchical testing procedure (ie, testing outcomes in a predefined order) was used for the primary and key secondary end points to ensure preservation of the overall type I error. All estimates used for the sample size determination were based on the results of the phase II study of abatacept in PsA,15 except for non-progressors using PsA-modified total SHS score. A two-sided continuity corrected χ2 test at alpha=0.05 was used. To achieve ≥80% power for each of the hierarchical end points and PASI 50 responders, recruitment of 400 patients was required: 152 (38%) and 248 (62%) in the TNFi-naïve and TNFi-exposed subgroups, respectively (see online section 3 in the supplementary appendix).
Supplemental material
All efficacy analyses (including those up to week 44 or 52) were conducted using the intent-to-treat (ITT) population, which comprised all randomised patients who received at least one dose of study medication. Comparisons between treatment arms were performed for the primary and key secondary end points, and PASI 50 responders at week 24, using a two-sided Cochran-Mantel-Haenszel χ2 test, stratified by current methotrexate use, prior TNFi use and plaque psoriasis involving ≥3% of BSA, at a 5% significance level for generating p values. The p values that did not control for overall type I error (nominal p values) were provided for end points that ranked lower in the statistical hierarchy than the first end point that was non-statistically significant at the 5% level, and for PASI 50 response, MDA and DAPSA score at week 24. For other end points, only 95% CIs of differences between abatacept and placebo arms were generated without obtaining p values. For binary responder analyses during the double-blind period, EE patients at week 16 were imputed as non-responders at weeks 20 and 24 (for radiographic analysis, EE patients were imputed as progressors at week 24). Patients who discontinued treatment were imputed as non-responders/radiographic progressors at all subsequent visits. Continuous variables for the double-blind analysis were assessed using a longitudinal repeated-measures analysis, imputing EE patients as missing beyond week 16 (see online section 3 in the supplementary appendix). In addition, if there were still missing data (for EE patients between week 4 and week 16 and for non-EE patients between week 4 and week 24), patients were imputed as non-responders at the time point with missing data. However, if data were missing between two time points at which the patient had a response (eg, ACR20), then in such cases the response (eg, ACR20) was imputed at the time point with missing data.
Analyses up to week 44/52 used actual data at each time point for EE and non-EE patients. A non-responder imputation was done for all missing values regardless of escape status. As mentioned above, the denominators for all responder analyses up to week 44/52 were equal to all randomised and treated patients (ITT population). Most efficacy end points are reported only up to week 44, at which time EE patients had received 28 weeks and non-EE patients had received 20 weeks of open-label treatment. However, for analyses of enthesitis, dactylitis and radiographic data, week 44 data from EE patients were combined with week 52 data from non-EE patients, as these data were not collected at week 44 for non-EE patients. Continuous variables were analysed for this period using the longitudinal repeated-measures analysis model using the actual data including all patients in the ITT population. For SHS scores, adjusted mean change from baseline up to week 44/52 was calculated using the longitudinal repeated measures analysis model with the actual values for EE and non-EE patients (ITT population).
Results
Patients
In total, 424 randomised patients received at least one dose of abatacept (n=213) or placebo (n=211). Patient characteristics at baseline are shown in table 1. The overall mean (SD) age was 50.4 (11.0) years, 55% were female and 60% reported current methotrexate use, with a mean (SD) dose of 17.1 (8.2) mg/week at baseline. Most patients (~60%) had previously received TNFi agents; of these, most (abatacept 60%, placebo 62%) had failed at least one TNFi due to inadequate efficacy. Overall, 69% of patients had psoriasis covering ≥3% of BSA. Numbers of non-biologic DMARDs used prior to study entry are described in online table 1 in the supplementary appendix. The baseline disease characteristics included mean (SD) disease duration of 8.5 (8.2) years; distal interphalangeal involvement in approximately half of the population (50.7%); presence of joint erosion on radiographs in 84% of patients, with a mean (SD) PsA-modified total SHS score of 18.8 (43.3); elevated serum CRP above upper limit of normal (3 mg/L) in 66% of patients, with a mean (SD) CRP of 14.1 (25.9) mg/L; and polyarticular disease in 98% of patients, with mean (SD) tender and swollen joint counts of 20.2 (13.3) and 11.6 (7.5), respectively.
Supplemental material
Patient characteristics at baseline
Patient disposition is shown in figure 1. A total of 76 (35.7%) and 89 (42.2%) patients in the abatacept and placebo groups, respectively, were assigned to EE and switched to open-label abatacept at week 16. From the original abatacept and placebo arms, 197 (92.5%) and 185 (87.7%) patients, respectively, entered the open-label period.
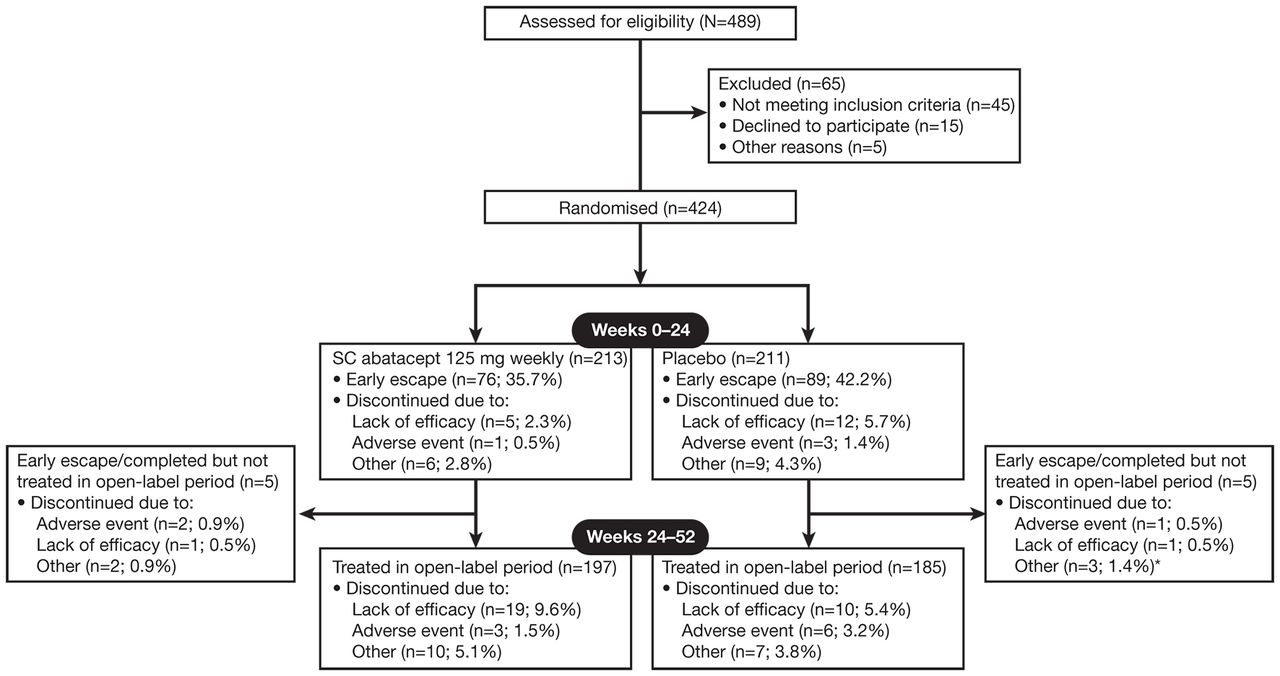
Patient disposition. SC, subcutaneous. *Includes missing (n=2).
Musculoskeletal manifestations
Arthritis
Abatacept treatment resulted in a significantly higher proportion of patients achieving an ACR20 response at week 24 versus placebo (39.4% vs 22.3%; p<0.001; table 2; figure 2; online table 2 in the supplementary appendix). The mean change from baseline in each of the ACR core components was numerically greater for patients in the abatacept group than those in the placebo group at 24 weeks (see online table 3 in the supplementary appendix).
Supplemental material
Supplemental material
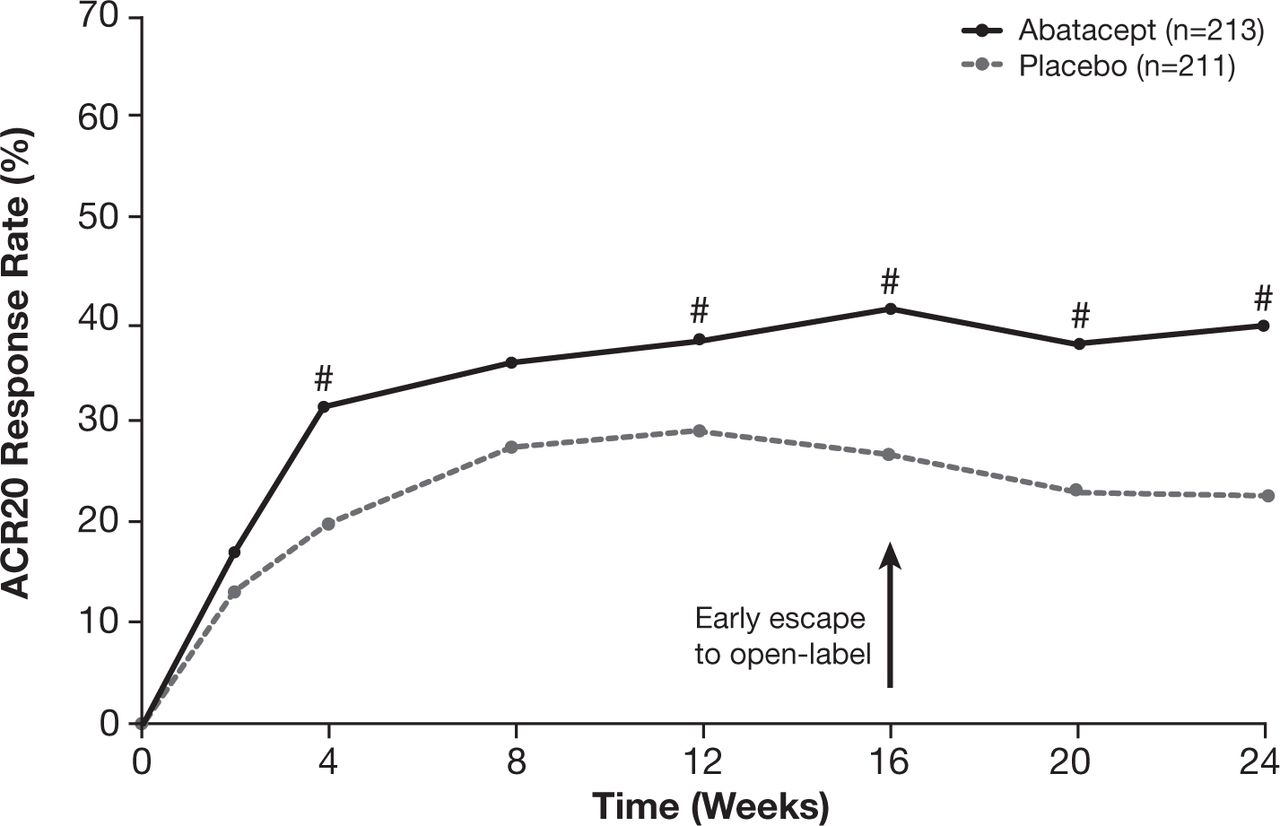
ACR20 response over the 24-week double-blind period (non-responder imputation for early escape). Early escape patients switching to open-label abatacept at week 16 were imputed as non-responders at weeks 20 and 24. If there were still missing data, patients were imputed as non-responders, unless data were missing between two time points at which the patient had a response, in which case response was imputed. #Where 95% CI of estimate of differences in ACR20 responses for abatacept versus placebo do not contain zero. ACR20, ≥20% improvement in the American College of Rheumatology criteria.
ACR20/50/70 responders in the total population and TNFi-naïve and TNFi-exposed subgroups (ITT population)
As the effect of abatacept on the first key secondary end point in the statistical hierarchy (HAQ-DI response rate) did not reach significance (see below), only nominal p values were generated for subsequent outcomes. Nominally higher ACR20 response rates with abatacept versus placebo were seen in both TNFi-naïve and TNFi-exposed subgroups at week 24 (table 2), with the largest treatment difference seen in TNFi-naïve patients. ACR20 responses at 24 weeks by number of prior TNFi received are shown in online table 4 in the supplementary appendix. Analysis (ITT population) up to week 44 showed that ACR20 responses were maintained for patients who continued abatacept and improved for those who switched from placebo to abatacept (placebo/abatacept) in the total population and in both TNFi-naïve and TNFi-exposed subgroups (table 2; figure 3; online table 5 in the supplementary appendix). Because the trial design allowed for early escape to open-label abatacept, improvement in the placebo-treated patients initiating active treatment would be expected starting at week 16. Similar trends were observed for ACR50 and ACR70 responses (table 2). In addition, patients with CRP elevated above the upper limit of normal at baseline showed the highest ACR20 responses at 24 weeks with abatacept treatment versus placebo (estimated differences (95% CI)): total population, 43.8% versus 23.7% (20.17 (9.32 to 31.02)); TNFi naïve, 50.0% versus 23.9% (26.09 (7.93 to 44.25)); TNFi exposed, 40.2% versus 23.5% (16.69 (3.21 to 30.17)).
Supplemental material
Supplemental material

{kind=link}
{kind=link}
{kind=link}
Proportion of patients achieving ACR20 response (ITT analysis, actual data for early escape patients) over the combined double-blind and open-label periods in the total population (A) and the TNFi-naïve (B) and TNFi-exposed (C) subgroups. Error bars represent 95% CIs. For EE patients, measurements at weeks 20, 24, 28, 36 and 44 are actual measurements at weeks 4, 8, 12, 20 and 28 of open-label abatacept treatment. The increase in the proportion of patients with ACR20 response from week 16 to week 24 in the placebo group reflects the mixed population of EE patients who received abatacept between weeks 16 and 24 and non-EE patients who received placebo at week 24. If there were missing data, patients were imputed as non-responders, unless data were missing between two time points at which the patient had a response, in which case response was imputed. ACR20, ≥20% improvement in the American College of Rheumatology criteria; EE, early escape; ITT, intent to treat; TNFi, tumour necrosis factor inhibitor.
The efficacy of abatacept in reducing arthritic manifestations was supported by the results of the post hoc analysis of greater improvement in DAS28 (CRP) from baseline to week 24 with abatacept versus placebo: adjusted mean change, –1.35 versus –0.94; adjusted difference (95% CI), –0.42 (–0.69 to –0.14). Continued improvement beyond week 24 in the ITT population was seen in adjusted mean changes from baseline in DAS28 (CRP) in both abatacept and placebo/abatacept groups, with changes from baseline to week 44 of –1.81 and –1.84, respectively (see online supplementary figure 1 and table 6 in the supplementary appendix).
Supplemental material
Supplemental material
Enthesitis and dactylitis
At week 24, complete resolution of enthesitis and dactylitis present at baseline was numerically more frequent with abatacept versus placebo. The proportions (95% CI) of patients with enthesitis resolution were 32.9% (25.1 to 40.6) versus 21.2% (14.2 to 28.2) and with dactylitis resolution were 44.3% (31.8 to 56.7) versus 34.0% (20.9 to 47.1), respectively. At week 44/52, an increased proportion of patients achieved complete resolution of baseline enthesitis (48.6% vs 43.9%) and dactylitis (68.9% vs 60.0%) in both the abatacept and placebo/abatacept groups, respectively.
Physical function
The proportion of patients with an HAQ-DI response (reduction from baseline score ≥0.35) at week 24 was numerically higher with abatacept versus placebo: 31.0% versus 23.7%; estimated difference (95% CI), 7.2 (–1.1 to 15.6); p=0.097. However, as this did not reach statistical significance, only nominal p values were generated for lower-ranking end points in the hierarchical testing. HAQ-DI responses at 24 weeks in the abatacept versus placebo arms were 34.5% versus 19.8%, respectively, in the TNFi-naïve subgroup (estimated difference 14.8; 95% CI 1.7 to 28.0) and 28.7% versus 26.2%, respectively, in the TNFi-exposed subgroup (estimated difference 2.5; 95% CI –8.3 to 13.3). HAQ-DI responses were maintained to week 44 in the abatacept group and improved in the placebo/abatacept group (39.9% and 38.9%, respectively) in the ITT population.
Further analyses showed nominal improvements in adjusted mean change in HAQ-DI score from baseline to week 24 with abatacept versus placebo for all patients: –0.33 versus –0.20, respectively; estimated difference (95% CI), –0.13 (–0.25 to –0.01), and in both TNFi-naïve (–0.29 vs –0.17) and TNFi-exposed (–0.35 vs –0.18) subgroups. Continued improvements were seen in the total population and TNFi-naïve and TNFi-exposed subgroups at week 44 in the ITT analyses (see online supplementary figure 2 and table 7 in the supplementary appendix).
Supplemental material
Supplemental material
Structural damage
The proportion of patients without radiographic progression at week 24 was 42.7% in the abatacept group versus 32.7% in the placebo group (estimated difference (95% CI), 10.0 (1.0 to 19.1); nominal p=0.034). The mean (SE) change from baseline in PsA-modified total SHS score was 0.30 (0.12) versus 0.35 (0.13) at week 24 for abatacept versus placebo and 0.18 (0.12) versus 0.30 (0.12) at week 44/52 for abatacept versus placebo/abatacept.
Psoriatic skin responses
The psoriatic skin response was more modest compared with the musculoskeletal response. At week 24, there was a small numerical increase in the proportion of PASI 50 responders with abatacept compared with placebo: 26.7% versus 19.6% (estimated difference (95% CI), 7.3 (–2.2 to 16.7); nominal p=0.137). The proportion of patients with ≥75% improvement in PASI score from baseline (PASI 75 responders) with abatacept versus placebo at week 24 was 16.4% versus 10.1%, respectively.
The magnitude of improvement in both PASI 50 and PASI 75 response rates with abatacept versus placebo at week 24 was numerically greater in the TNFi-naïve compared with the TNFi-exposed subgroup: PASI 50 (TNF naïve, 32.7% vs 19.6%; TNFi exposed, 23.1% vs 19.6%) and PASI 75 (TNFi naïve, 18.2% vs 9.8%; TNFi exposed, 16.5% vs 10.3%).
In the ITT population at week 44, PASI 50 response rates were maintained for patients who continued on abatacept (total population, 30.1%; TNFi naïve, 36.4%; TNFi exposed, 26.4%) and improved for those who switched from placebo to abatacept (total population, 34.5%; TNFi naïve, 39.2%; TNFi exposed, 32.0%). PASI 75 responses were also maintained for patients who continued on abatacept (total population, 19.9%; TNFi naïve, 27.3%; TNFi exposed, 15.4%) and improved for the placebo/abatacept group (total population, 16.9%; TNFi naïve, 17.6%; TNFi exposed, 16.5%).
Disease activity—composite measures
The proportion of patients with MDA at week 24 was numerically higher with abatacept versus placebo in the total population (11.7% vs 8.1%; nominal p=0.205). At week 52, the proportion of patients with MDA increased to 17.4% for patients who continued on abatacept and 18.5% for the placebo/abatacept group. Similar trends were observed in the modified Composite Psoriatic Disease Activity Index and Psoriatic Arthritis Disease Activity Score (see online section 4 in the supplementary appendix). There was a nominally significant improvement with abatacept versus placebo in adjusted mean change from baseline to week 24 in DAPSA score (–18.75 vs –13.00; adjusted difference –5.75; 95% CI –10.01 to –1.49; nominal p=0.008). At week 44, further improvements in adjusted mean change from baseline in DAPSA score were observed in the abatacept and placebo/abatacept groups (–24.58 and –25.18, respectively).
Supplemental material
Quality of life
At 24 weeks, mean improvements from baseline were numerically greater with abatacept versus placebo for SF-36 physical component summary and Dermatology Life Quality Index scores but were similar between the two groups for SF-36 mental component summary scores. Similar results were also seen at week 52 (see online table 8 in the supplementary appendix).
Supplemental material
Safety
Safety findings during the 24-week, double-blind period and for cumulative abatacept treatment over the 52-week study period are summarised in table 3. During the 24-week, double-blind period, the abatacept and placebo groups had similar safety profiles, with comparable incidences, respectively, of serious AEs (2.8% vs 4.3%), AEs (54.5% vs 53.1%) and infections (26.8% vs 29.9%). One serious infection (Pneumocystis jirovecii) was considered related to study drug by the investigator and led to treatment discontinuation. This event occurred during the double-blind period in a patient receiving abatacept who had a history of smoking and chronic obstructive pulmonary disease and had recently used high-dose corticosteroids. The episode resolved after 7 days of appropriate treatment.
Summary of safety
Discussion
In this phase III study, selective modulation of T-cell costimulation with abatacept resulted in significantly higher ACR20 response rates in patients with PsA compared with placebo, with responses maintained to at least 1 year. Our findings support previous data suggesting a role for T cells in PsA: activated T cells are abundant in the synovial fluid of patients with PsA31 32 and frequencies of interleukin (IL)-17-secreting CD8+ T cells are increased in erosive disease.32 Furthermore, treatment with abatacept has been shown to reduce circulating IL-17-secreting CD4+ and CD8+ T cells in RA.33 The data reported here suggest that selective inhibition of the CD28-dependent costimulatory pathway critical for T-cell activation7 may offer a novel treatment option in PsA.
Our findings demonstrate that abatacept had an overall beneficial effect on musculoskeletal symptoms and was well tolerated in a relatively refractory population of patients with PsA (approximately 60% had received prior TNFi), confirming earlier results from a phase II, dose-ranging study in a less refractory population (approximately 30% of patients in the abatacept 10 mg/kg and placebo groups had received prior TNFi).15 The primary end point was met, with a statistically significantly higher ACR20 response at 24 weeks with abatacept treatment versus placebo. Although numerical improvements in individual ACR core components were observed with abatacept versus placebo at 24 weeks, the CIs were overlapping. Due to the lack of significant effect on HAQ-DI response rates in the total population, it was not possible to attribute significance to lower-ranking outcomes in the statistical hierarchy. The efficacy in joints was supported by mean improvements in DAS28 (CRP) with abatacept versus placebo. In addition, disease improvement was evident when placebo-treated patients switched to abatacept. Outcomes tended to be better in the TNFi-naïve versus TNFi-exposed subgroups.
Across end points up to week 52, responses were maintained or improved for patients who continued on abatacept, demonstrating the durability of effects and accrual of benefits over time on some measures. For patients who switched from placebo to open-label abatacept, it is possible that observed improvements could at least partially be explained by patient awareness of receiving active treatment, or a continuation of trends during receipt of placebo, rather than a true treatment effect. However, the similar ACR20 response rates at 44 weeks for patients who started on abatacept and those who switched from placebo to abatacept indicate a treatment effect and benefit after switching.
Skin responses to abatacept were modest. A small treatment effect on skin manifestations has previously been observed with the T-cell inhibitor, alefacept, in a phase III study in psoriasis.34 It is also possible that a higher dose of abatacept may be required for optimal efficacy in skin versus musculoskeletal symptoms, similar to previous findings with the TNFi etanercept.35
Caution is advised when comparing the current efficacy data with findings from studies of TNFi and other agents in PsA. The ACR20 response rate at week 24 in this study was lower than that in previous studies of agents that target some of the known effector molecules in PsA.36–38 However, this study included a higher proportion of TNFi failures compared with most studies,36 37 which may indicate a more treatment-refractory population, as noted previously.39 Higher efficacy in the TNF-naive compared with the TNFi-exposed subgroup across multiple end points in the current study confirmed the treatment resistance in the latter subpopulation. In contrast to findings with other agents with different mechanisms of action,36–38 abatacept treatment demonstrated better efficacy on musculoskeletal versus skin end points. The reasons for this are unclear but may include differential dose requirements for optimal efficacy of abatacept in skin versus the joints, for example, due to less efficient drug penetration of skin versus synovial tissue, and distinct pathologies with divergent roles of T cells and T-cell subsets in skin versus synovial inflammation in PsA. Regarding the latter, it is interesting to note that, in PsA, agents targeting the IL-23/IL-17 axis can achieve complete clearing of psoriatic skin lesions without a similar level of efficacy in the joints.40 We speculate that T-cell subsets driving pathology in the skin and joints may differ in their expression of CD28 and, hence, susceptibility to abatacept.
In this study, subcutaneous abatacept was well tolerated with no new safety signals, consistent with the phase II study of intravenous abatacept in PsA15 and previous studies of subcutaneous and intravenous abatacept in RA.41 Throughout the study, one serious opportunistic infection was reported. This case of P. jirovecii infection occurred in the abatacept arm in a patient who had a history of smoking and chronic obstructive pulmonary disease and had recently received high doses of corticosteroids. It has been recognised that patients with chronic obstructive pulmonary disease have an increased prevalence of Pneumocystis colonisation, which may predispose them to acute infection.42
Limitations of this study include the prespecified imputation method used to measure radiographic progression. Imputation as radiographic progressors of patients who escaped early to open-label abatacept, based on poor clinical response at week 16, led to a relatively high imputed rate of structural progression at week 24 in both groups. This imputation method that was designed initially is inappropriate as it assumed that the structural radiographic data behaved similarly to clinical data and obscured underlying rates of radiographic change. Overall, there was minimal progression based on the mean change from baseline in PsA-modified total SHS score over 24 and 44/52 weeks in both groups, making it difficult to detect meaningful treatment differences. In this context, it should be noted that, in the phase II study in PsA, abatacept demonstrated greater inhibition of structural damage versus placebo as well as improvements in joint inflammation on MRI over the same timeframe (24 weeks).15
In summary, abatacept treatment achieved the primary end point in ACR20 response rates in patients with PsA, of whom ~60% had prior exposure to TNFi agents. There were trends towards benefits in other musculoskeletal measures, with maximal effects seen in the TNFi-naïve patients. However, only modest benefit was demonstrated for psoriatic skin lesions. Abatacept was well tolerated with no new safety signals.
References
Footnotes
Contributors PM, ABG and DG were involved in the conception and design of the study and interpretation of data. DvdH and MN were involved in the conception and design of the study, acquisition of data and interpretation of data. OF was involved in interpretation of data. SB was involved in the acquisition of data and the analysis and interpretation of data. AJ was involved in the conception and design of the study, acquisition of data and the analysis and interpretation of data. All authors had full access to the study data, critically reviewed the manuscript and approved the final version prior to submission and take responsibility for the integrity and accuracy of the reported data.
Funding This study was funded by Bristol-Myers Squibb. Under the direction of the authors, Sharon Gladwin of Caudex, Oxford, UK (funded by Bristol-Myers Squibb) provided writing assistance for the development of this manuscript. Editorial assistance was provided by Paul Wilmott of Caudex, funded by Bristol-Myers Squibb. Sandra Overfield, Protocol Manager, Bristol-Myers Squibb assisted in initial design and operational aspects of the study.
Competing interests PM reports receiving consulting fees from AbbVie, Amgen, Bristol-Myers Squibb, Celgene, Corrona, Crescendo, Demira, Janssen, Lilly, Merck, Novartis, Pfizer, Sun, UCB and Zynerba; and speaker fees from AbbVie, Amgen, Bristol-Myers Squibb, Celgene, Crescendo, Genentech, Janssen, Novartis, Pfizer and UCB. ABG reports receiving consulting fees from Abbott Laboratories (AbbVie), Actelion, Akros, Amgen, Astellas, Baxalta, Beiersdorf, Bristol-Myers Squibb, Canfite, Catabasis, Celgene, Centocor (Janssen), Coronado, CSL Behring Biotherapies for Life, Dermipsor, Genentech, GlaxoSmithKline, Incyte, Karyopharm, Kineta One, KPI Therapeutics, Lilly, Meiji Seika Pharma, Mitsubishi Tanabe Pharma Development America, Novartis, Novo Nordisk, Pfizer, Takeda, TEVA, UCB, Vertex and Xenoport; and research grants (paid to Tufts Medical Center) from Abbott Laboratories (AbbVie), Amgen, Baxalta, Celgene, Centocor (Janssen), Dermira, Levia, Lilly, Merck, Novartis, Pfizer and Xenoport. DvdH reports receiving consultancy fees from Bristol-Myers Squibb and also consultancy fees from AbbVie, Amgen, Astellas, AstraZeneca, Boehringer Ingelheim, Celgene, Daiichi, Eli Lilly, Galapagos, Gilead, Janssen, Merck, Novartis, Pfizer, Regeneron, Roche, Sanofi and UCB (outside the submitted work). OF reports receiving grant support from AbbVie, Bristol-Myers Squibb and Pfizer; and speaker fees from Celgene, Janssen, Novartis and UCB. AJ is an employee of Bristol-Myers Squibb and reports holding stock in Bristol-Myers Squibb. MN is an employee of Bristol-Myers Squibb. SB is an employee of Bristol-Myers Squibb and reports holding stock in Bristol-Myers Squibb. DG reports receiving grant support from Bristol-Myers Squibb to participate in this study; in addition, she reports receiving grant support and fees from AbbVie, Amgen, Celgene, Eli Lilly, Janssen, Novartis, Pfizer and UCB.
Ethics approval Schulman Associates Institutional Review Board.
Provenance and peer review Not commissioned; externally peer reviewed.