Article Text

Abstract
Objectives To compare efficacy and safety of various doses of tofacitinib, an oral Janus kinase inhibitor, with placebo in patients with active ankylosing spondylitis (AS, radiographic axial spondyloarthritis).
Methods In this 16-week (12-week treatment, 4-week washout), phase II, multicentre, dose-ranging trial, adult patients with active AS were randomised (N=51, 52, 52, 52, respectively) to placebo or tofacitinib 2, 5 or 10 mg twice daily. The primary efficacy endpoint was Assessment of SpondyloArthritis International Society 20% improvement (ASAS20) response rate at week 12. Secondary endpoints included objective measures of disease activity, patient-reported outcomes and MRI of sacroiliac joints and spine. Safety was monitored.
Results Emax model analysis of the primary endpoint predicted a tofacitinib 10 mg twice daily ASAS20 response rate of 67.4%, 27.3% higher than placebo. Supportive normal approximation analysis demonstrated tofacitinib 5 mg twice daily ASAS20 response rate significantly higher than placebo (80.8% vs 41.2%; p<0.001); tofacitinib 2 and 10 mg twice daily demonstrated greater response rate than placebo (51.9% and 55.8%, respectively; not significant). Secondary endpoints generally demonstrated greater improvements with tofacitinib 5 and 10 mg twice daily than placebo. Objective (including MRI) endpoints demonstrated clear dose response. Adverse events were similar across treatment groups with no unexpected safety findings. Dose-dependent laboratory outcome changes returned close to baseline by week 16.
Conclusions Tofacitinib 5 and 10 mg twice daily demonstrated greater clinical efficacy versus placebo in reducing signs, symptoms and objective endpoints of active AS in adult patients with a similar 12-week safety profile as reported in other indications.
Trial registration number NCT01786668.
- Ankylosing Spondylitis
- Magnetic Resonance Imaging
- Disease Activity
- Treatment
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Ankylosing spondylitis (AS), also called radiographic axial spondyloarthritis (axSpA), is a chronic systemic inflammatory disease of the axial skeleton with major impact on quality of life.1 The management of AS comprises physical therapy alongside non-steroidal anti-inflammatory drugs (NSAIDs), followed by tumour necrosis factor inhibitors (TNFi) for persistent disease.2–5 No evidence exists for efficacy of conventional synthetic disease-modifying antirheumatic drugs (DMARDs) for treating axial disease.3 Recently, secukinumab, an interleukin (IL)-17 blocker, became available as a treatment for AS,6 but treatment options remain limited.6 ,7 Therefore, an unmet need exists for therapies with alternative mechanisms of action to control and manage radiographic axSpA.8
Tofacitinib is an oral Janus kinase (JAK) inhibitor. Through direct and indirect inhibition of cytokine pathways, tofacitinib can modulate immune responses and reduce or prevent inflammation.9 In cellular settings, tofacitinib preferentially inhibits signalling via JAK3 and/or JAK1 with functional selectivity over signalling via pairs of JAK2.10 ,11 This affects signalling via IL-17, IL-21 and IL-23,9 ,12 which have been implicated in AS pathology13–15 and antibodies to IL-17 have demonstrated efficacy in AS.6 ,16 In addition, tofacitinib reduced serum levels of TNF9 and TNFi have been found to be efficacious in the treatment of active AS.3 Extra-articular manifestations associated with AS include inflammatory diseases such as psoriasis, inflammatory bowel disease and uveitis.17 The efficacy and safety of tofacitinib have been studied in several immune-mediated inflammatory diseases, such as rheumatoid arthritis,18–23 psoriasis24–26 and ulcerative colitis.27 ,28 Results of the first proof-of-concept dose-ranging investigation of the effects of tofacitinib in adult patients with active AS are reported. This study also represents the first dose-ranging study in AS.
Methods
This was a 16-week (12-week treatment, 4-week washout), phase II, multicentre, randomised, double-blind, placebo-controlled, dose-ranging study (NCT01786668) to investigate the efficacy, safety and dose response of tofacitinib in patients with active AS.
Patients
Patients entering the study were ≥18 years of age and fulfilled the modified New York (mNY) criteria for AS, confirmed by centralised reading of sacroiliac (SI) radiographs. Patients had active disease based on Bath AS Disease Activity Index (BASDAI) score ≥4 and back pain score ≥4 and history of either inadequate response to ≥2 oral NSAIDs or intolerance to prior NSAIDs. Patients with C reactive protein (CRP) levels within the normal reference range and those with active arthritis, enthesitis or psoriasis could be enrolled, provided mNY criteria for AS were met. Patients were permitted to continue concurrent treatment with methotrexate, sulfasalazine and stable oral corticosteroids (<10 mg/day of prednisone or equivalent). No eligibility criteria relating to MRI were specified. Exclusion criteria (see online supplementary section 1) included current or prior biological DMARD treatment and evidence of active, latent or inadequately treated tuberculosis infection.
supplementary data
Patients were recruited at 58 centres globally (see online supplementary section 2). The study was conducted in accordance with applicable legal and regulatory requirements, and the general principles set forth in the International Ethical Guidelines for Biomedical Research Involving Human Subjects, International Conference on Harmonisation Guidelines for Good Clinical Practice and the Declaration of Helsinki. All patients provided written informed consent. Institutional review boards or independent ethics committees at each investigational centre approved the study.
Study design and treatments
Patients were randomised (1:1:1:1) to placebo or tofacitinib 2, 5 or 10 mg twice daily for 12 weeks plus a 4-week off-treatment follow-up period. Tofacitinib was dispensed in combinations of 1 and 5 mg tablets, with matching placebos, and administered orally.
Study endpoints
The primary efficacy endpoint was Assessment of SpondyloArthritis International Society 20% improvement (ASAS20) response rate at week 12. Secondary efficacy endpoints included ASAS20 at other time points, ASAS40, ASAS5/6, Ankylosing Spondylitis Disease Activity Score (ASDAS) with CRP major response (improvement ≥2.0) and clinical improvement (≥1.1), BASDAI50 and ASAS5/6 response rates, change from baseline in ASDAS, Bath AS Functional Index (BASFI) and Bath AS Metrology Index (linear method; BASMI), Maastricht AS Enthesitis Score (MASES), swollen joint counts, chest expansion and spinal mobility. MRI outcomes included week 12 change from baseline in SPondyloArthritis Research Consortium of Canada (SPARCC) score of SI joints and spine (six most severely affected discovertebral units), and Berlin modification of AS spine MRI score (Berlin score). Patient-reported outcomes (PROs) were assessed, including: AS Quality of Life (ASQoL), EuroQol 5 dimensions (EQ-5D), short-form 36 V2 (SF-36), functional assessment of chronic illness therapy—fatigue (FACIT-F) and Work Productivity and Activity Impairment. All endpoints except MRI were assessed at baseline and at weeks 2, 4, 8 and 12. MRI was performed after screening and within 7 days prior to baseline visit and during week 12 but prior to the study visit, or at early termination (if after study week 6). MRI scoring was conducted by two central readers, independently blinded to time sequence and treatment. The average score was used for analysis of continuous MRI endpoints; discrepancies between readers for binary endpoints were resolved via MRI scoring by a third trained adjudicator, blinded to the results of the two readers and visit.
Safety was monitored throughout the study, including adverse event (AE) reporting and laboratory outcomes.
Statistical analysis
The primary endpoint, ASAS20 response rate at week 12, was analysed by a prespecified three-parameter Bayesian Emax model (see online supplementary section 3) to characterise the dose–response relationship using the full analysis set (all patients receiving ≥1 dose of study drug). A supplementary analysis of the primary endpoint used the normal approximation to compare each tofacitinib dose with placebo. Non-responder imputation (NRI) was used for missing ASAS20 responses.
Sample size was determined by clinical trial simulations using the Emax model to estimate true ASAS20 responders at week 12 with different doses. Simulations were conducted under several plausible truths, assuming 50 patients per treatment group (200 in total). If the true placebo-corrected ASAS20 response in the range 1–10 mg twice daily was 20%–40%, the estimated placebo-corrected effect for that dose with limits ±10% was projected to capture the true placebo-corrected response rate ≥83% of the time.
Binary secondary endpoints were analysed using the normal approximation. Continuous endpoints were analysed as change from baseline with a marginal repeated measure model including treatment group, visit, treatment group-by-visit interaction and baseline value as fixed effects.
Safety data were summarised descriptively.
Subanalyses
Post hoc subanalyses were performed to identify whether baseline CRP or MRI status impacted efficacy (ASAS20 and ASAS40; online supplementary section 4).
Results
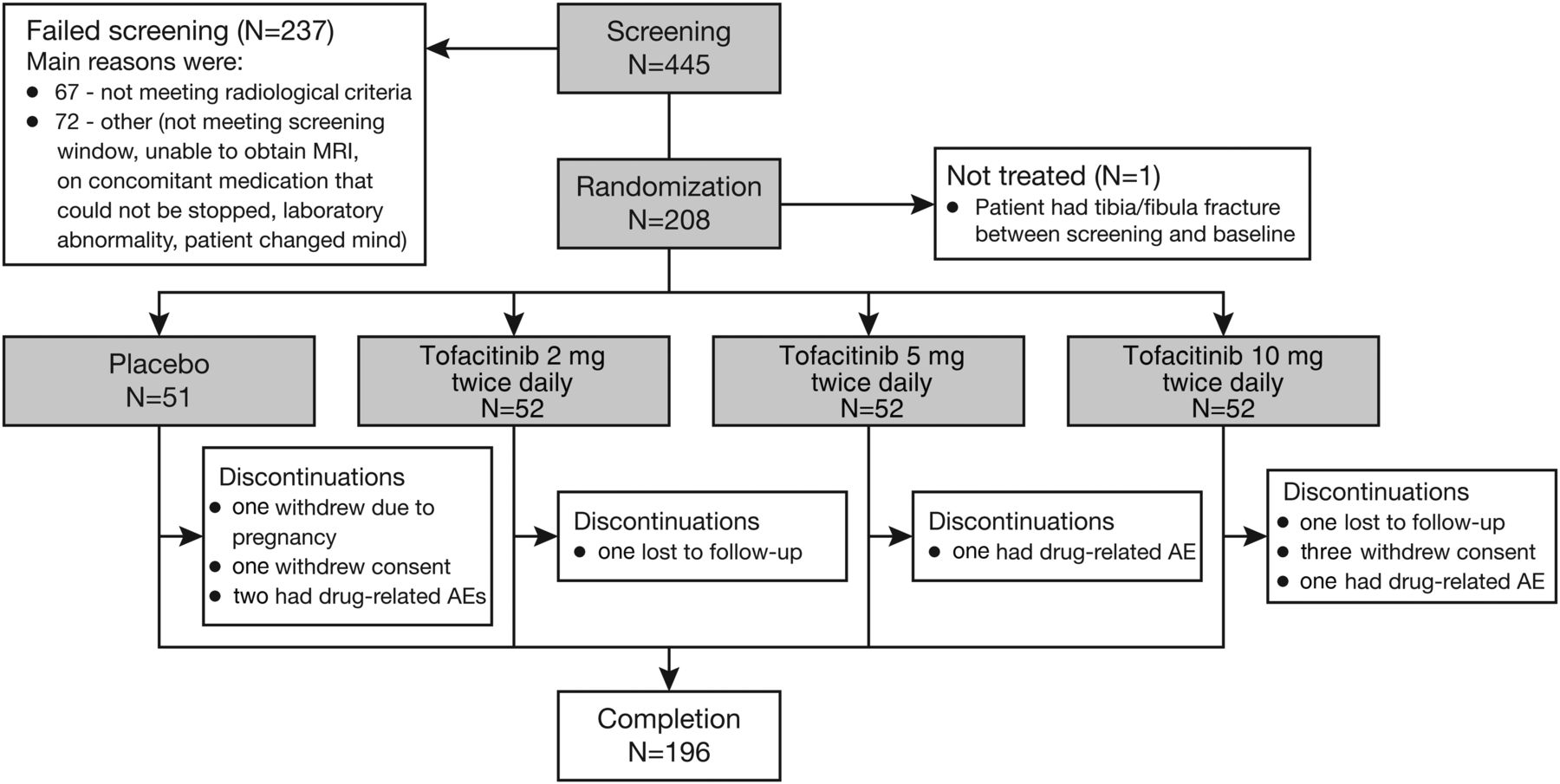
Between April 2013 and March 2015, 445 patients were screened, with 208 patients randomised and 207 treated (1 patient randomised to placebo did not receive study medication; figure 1). Of these, 196 patients completed the study; 51 placebo-treated patients and 52 in each tofacitinib group were included in analyses (figure 1). Of the 237 patients who failed screening, 67 patients did not meet radiological criteria (figure 1).

Patient disposition. AE, adverse event.
Baseline demographics and disease characteristics were generally similar between groups and typical of an active AS population (table 1).
Baseline demographics and disease characteristics
ASAS20 response rates using NRI at week 12 (primary endpoint) for placebo and tofacitinib 2, 5 and 10 mg twice daily are shown in table 2. According to the Emax model, the predicted 10 mg dose response rate was 67.4%; 11.4% and 27.3% higher than the tofacitinib 2 mg twice daily and placebo rates, respectively. As per normal approximation for comparing active treatment with placebo, response rates were significantly greater with tofacitinib 5 mg twice daily versus placebo (p<0.001), but not with tofacitinib 2 or 10 mg twice daily.
Primary efficacy endpoint results: ASAS20 response rate at week 12
Secondary and other efficacy endpoint responses at week 12 are presented in table 3. All tofacitinib groups had ASAS40 and BASDAI50 response rates and change in ASDAS of similar magnitude, and were significant versus placebo. All tofacitinib groups had statistically significant improvement from baseline in BASDAI versus placebo. ASAS5/6 response rates were significantly greater with tofacitinib 5 and 10 mg twice daily versus placebo. ASDAS clinically important improvement response rates were significantly higher in all tofacitinib groups versus placebo; ASDAS major response was not significantly different between tofacitinib and placebo. ASDAS low/moderate disease was significantly higher with tofacitinib 2 and 5 mg twice daily versus placebo while there was no significant difference between treatment groups for ASDAS inactive disease. Tofacitinib 5 and 10 mg twice daily demonstrated significant improvement versus placebo in SPARCC SI joint scores, while all tofacitinib doses had significant improvement versus placebo in SPARCC spine scores (figure 2). Tofacitinib 5 and 10 mg twice daily demonstrated significant improvement versus placebo in Berlin scores (−2.2 and −2.13 vs −0.41; p<0.001 and p=0.001, respectively). Mean and mean change from baseline CRP values at week 12 were similar across all groups.
Secondary and other efficacy endpoint responses at week 12

{kind=link}
{kind=link}
Secondary clinical efficacy endpoints by study visit: (A) mean (SE) Ankylosing Spondylitis Disease Activity Score (ASDAS) major improvement; least squares (LS) mean (SE) change from baseline in (B) Bath Ankylosing Spondylitis Functional Index (BASFI) and (C) Bath Ankylosing Spondylitis Metrology Index (BASMI); LS mean (SE) change from baseline to week 12 in SPondyloArthritis Research Consortium of Canada (SPARCC) scores of (D) sacroiliac (SI) joints and (E) spine; and cumulative probability plots of change in SPARCC score of (F) SI joints and (G) spine.
Secondary efficacy endpoints at other study visits are presented in online supplementary table S1 or figure 2 (including week 12 data). ASAS20 response rate over time is presented in online supplementary figure S1. Numerical separation from placebo for all tofacitinib doses was observed for all endpoints by week 4, except ASDAS inactive disease, where the response rate was small for all groups (<10%).
In the subset of study patients with enthesitis at baseline, tofacitinib 5 and 10 mg twice daily demonstrated statistically significant improvement versus placebo at week 12 in change from baseline in MASES (see online supplementary figure S2).
Mean swollen joint counts and mean chest expansion at each visit were similar across all groups (see online supplementary table S2).
Patients receiving tofacitinib experienced greater improvements versus placebo in EQ-5D mobility, SF-36 physical component summary and bodily pain, FACIT-F, ASQoL and work impairment (see online supplementary table S3).
Subanalyses demonstrated that patients with high (≥0.287 mg/dL (the upper limit of normal in the central laboratory)) baseline CRP had a larger treatment effect with tofacitinib versus placebo than those with low (<0.287 mg/dL) baseline CRP (ASAS20 and ASAS40; see online supplementary table S4). This is especially true for ASAS40 and mainly due to a lower placebo response in patients with high baseline CRP. For patients with positive baseline MRI (SPARCC SI joint cut-off ≥2 at baseline), greater treatment effect was observed with all doses of tofacitinib versus placebo; significant for tofacitinib 5 mg twice daily (ASAS20 87.5 vs 40.0 tofacitinib 5 mg twice daily vs placebo (p<0.001); ASAS40 48.4 vs 20.0 (p<0.01)) (see online supplementary table S5). Analysis of treatment effect (ASAS20 and ASAS40) by composite baseline CRP/MRI status demonstrated that tofacitinib-treated patients in the high CRP/MRI-positive subgroup had greater treatment effect at week 12 versus placebo compared with those who had neither (see online supplementary table S6). However, these data should be interpreted with caution due to the small patient numbers in the various subgroups.
Table 4 presents a safety summary. More patients reported treatment-emergent AEs (TEAEs) with tofacitinib 5 and 10 mg twice daily than tofacitinib 2 mg twice daily and placebo (53.8% and 51.9% vs 44.2% and 43.1%, respectively; online supplementary table S7). Overall, the most frequently reported TEAEs were nasopharyngitis (n=13) and upper respiratory tract infection (n=8). One cardiovascular AE (hypertension) was reported with tofacitinib 10 mg twice daily. Two treatment-related herpes zoster cases were reported (one each with tofacitinib 2 and 10 mg twice daily; online supplementary section 4). One serious infection event of chronic iridocyclitis (uveitis) was reported (tofacitinib 5 mg twice daily); however, an infective process was not confirmed. This was considered to be a chronic event unrelated to study treatment and resolved following treatment with intravenous antibiotics and glucocorticoids. No other serious infections or cases of tuberculosis, malignancy, gastrointestinal perforation or death were reported.
Summary of AEs and laboratory outcomes meeting monitoring criteria
Dose-dependent changes in laboratory outcomes were observed and returned to approximately baseline values by week 16, following the 4-week washout period. Only one patient had laboratory values meeting discontinuation criteria: a confirmed pregnancy in the placebo group.
Discussion
This was the first proof-of-concept dose-ranging study of tofacitinib in patients with AS and the first dose-ranging study in AS. A noteworthy aspect of this study was the use of central reading to confirm radiographic sacroiliitis as an entry criterion for the first time in a clinical trial of AS; therefore, it represents the most homogenous patient population with AS in a randomised clinical study to date. Approximately 15% of patients considered suitable for entry into this study by local investigator failed screening due to not meeting the radiological criteria after central read. This highlights the variation of radiological scoring by individual study investigators and suggests that central reading could benefit future study designs to ensure consistency in patients included based on radiographic sacroiliitis.
An Emax model was used to estimate the dose–response relationship for the ASAS20 response rate. The modelling predicted that at week 12, tofacitinib 5 and 10 mg twice daily had ASAS20 response rates of 63.0% and 67.4%, respectively. Normal approximation analysis demonstrated that tofacitinib 5 mg twice daily was the only tofacitinib dose statistically superior to placebo for the ASAS20 response rate at week 12; this could reflect sampling variability. Tofacitinib 10 mg twice daily was numerically better than placebo but the difference was not statistically significant by normal approximation. Results based on such a model in a phase II proof-of-concept study are used during clinical development of an investigational product in relation to the a priori go/no go decision to initiate a phase III study.
Overall efficacy data, excluding ASAS20, generally demonstrated significant improvement with tofacitinib 5 and 10 mg twice daily versus placebo over 12 weeks of treatment across most endpoints assessed, with minimal clinical difference between the two tofacitinib doses. Tofacitinib 2 mg twice daily also demonstrated significantly superior efficacy versus placebo in ASAS40 and ASDAS endpoints, except ASAS5/6. Separation from placebo was observed by week 8 in most secondary endpoints, except ASAS20, ASAS40 and ASAS5/6, which separated at week 4. This appears to suggest slower onset of efficacy compared with TNFi.29–32 However, comparing data across studies is challenging due to differences in patient populations and study design. Absolute ASAS20, ASAS5/6, ASAS40 and BASDAI50 week 12 response rates for tofacitinib doses in this study appear generally similar to those reported for TNFi in phase III studies in AS populations, although these studies reported lower placebo responses than we observed, particularly for ASAS20 and BASDAI50.30–32
The placebo response observed in this study was high; the reason for this is currently unknown, but does not appear to relate to differences in gender or geographical region. It is known that male patients with AS often experience better improvement in outcomes versus females; however, fewer male patients were enrolled in the placebo group compared with the tofacitinib groups. ASAS responses appeared consistent across geographical regions (data not shown), although small patient numbers in some groups limit any interpretation. One potential explanation may be the inclusion of patients with no objective signs of inflammation, for example, patients could be enrolled if they had normal CRP levels; however, this accounted for few patients.
Subanalyses by baseline CRP/MRI status indicated that patients with high baseline CRP had a better response with tofacitinib versus placebo than those with low CRP. In this study, the 5 mg twice daily arm had the highest number of patients with high CRP and baseline MRI inflammation. In the composite analysis, high CRP/positive MRI at baseline were associated with higher ASAS response rates in all tofacitinib groups versus placebo than subgroups with lower baseline CRP/negative MRI. Similar results have been reported for TNFi in an AS cohort, where high baseline CRP was a predictor of better treatment response.33 Moreover, a greater treatment effect in patients with positive MRI and/or elevated CRP is especially seen in studies of TNFi in patients with non-radiographic axSpA,34 ,35 while there are only limited data in patients with AS.36
Overall, we found that objective measures, such as SPARCC SI joint and spine scores and BASMI, demonstrated a dose response, while more subjective endpoints, including PROs, did not. Subjective endpoints can be influenced by many factors affecting the patient, not all of which may be drug related. It is also possible that subjective improvement in symptoms lags behind improvement in objective assessments. It may also be that ASAS20 is not sufficiently discriminatory and therefore no longer represents the preferred endpoint for assessing treatment effect, especially given the high placebo response, which is also observed in other trials.37 ,38 Other endpoints, such as ASDAS or ASAS40, may be more suitable as primary outcome measures. However, a combination of assessments, including subjective and objective measures such as imaging, may be needed to evaluate overall disease improvement in early development.
The pattern of AEs and changes in laboratory outcomes were similar to those reported in previous tofacitinib studies in other indications.18–27 ,39–44 Laboratory test outcomes returned to approximately baseline values by week 16, following the 4-week washout period, which is also consistent with observations in other indications and may be important if patients require treatment withdrawal.
One limitation of our study was the 12-week duration of active treatment. The duration was chosen based on onset of efficacy observed in previous tofacitinib clinical trials in other indications, while balancing patient safety with a limited exposure time. It is possible that a maximum efficacy response was not reached by week 12.
Small sample sizes per arm may have contributed to the lack of a dose response in many of the subjective endpoints. Small sample sizes per arm also made potentially interesting subgroup analyses difficult to interpret. Although patients were required to have a prespecified level of disease activity at enrolment, they were not required to have a prespecified level of CRP (inflammatory activity) to enter the trial, which may have impacted the results. However, this study captured a population typical of AS and trials of TNFi in AS have generally not required high CRP at screening.29 ,31 ,32
In summary, this is the first study to evaluate the efficacy and safety of a JAK inhibitor in AS. Tofacitinib 5 and 10 mg twice daily demonstrated greater clinical efficacy versus placebo in reducing the signs, symptoms and spinal inflammation of AS in adults with active disease. The 12-week safety profile was similar to that reported for tofacitinib studies in other indications and no new safety signals or concerns were identified. Our results suggest that JAK inhibition may present a new mode of action for managing AS and could add to the currently limited treatment options;2 ,7 ,8 however, other trials are needed to adequately evaluate the treatment effect of JAK inhibitors in AS.
Acknowledgments
The authors thank the patients and investigators for their participation in this study (see online supplementary section 2).
References
Footnotes
Handling editor Tore K Kvien
Contributors DvdH, DF, TH, KSK and SM contributed to the study design. DF, DvdH, DL, TH, SM and KSK contributed to data analysis. All authors contributed to data interpretation, and planning and critical review of the manuscript content. All authors were responsible for the final decision to submit. Medical writing support, under the direction of the authors, was provided by Amanda Pedder of Complete Medical Communications.
Funding This study was sponsored by Pfizer Inc. Medical writing support, under the direction of the authors, was provided by Amanda Pedder of Complete Medical Communications and funded by Pfizer Inc.
Competing interests DvdH is a consultant for AbbVie, Amgen, AstraZeneca, Augurex, BMS, Boehringer Ingelheim, Celgene, Centocor, Chugai, Covagen, Daiichi, Eli Lilly, Galapagos, GSK, Janssen Biologics, Merck, Novartis, Novo Nordisk, Otsuka, Pfizer, Roche, Sanofi-Aventis, UCB and Vertex; and is director of Imaging Rheumatology. AD has received research grants from AbbVie, Amgen, Eli Lilly, Janssen, Novartis, Pfizer and UCB, and has been an advisory board member for AbbVie, Amgen, Boehringer Ingelheim, Janssen, Novartis, Pfizer and UCB. JCW has received research grants or consultation fees from AbbVie, BMS, Celgene, Chugai, Eisai, Janssen, Novartis, Pfizer, Sanofi-Aventis, TSH Taiwan and UCB. ED has participated in clinical trials sponsored by AbbVie, BMS, Celgene, Pfizer, Novartis, UCB, Amgen, Lilly and Sanofi-Aventis. KSK, DF, TH, DL and SM are employees and shareholders of Pfizer, Inc.
Ethics approval The study protocol and informed consent documentation were approved by institutional review boards or independent ethics committees at each investigational centre.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement All authors had full access to study data and had final responsibility for the decision to submit.