Article Text

Abstract
Background: Mikulicz’s disease (MD) has been considered as one manifestation of Sjögren’s syndrome (SS). Recently, it has also been considered as an IgG4-related disorder.
Objective: To determine the differences between IgG4-related disorders including MD and SS.
Methods: A study was undertaken to investigate patients with MD and IgG4-related disorders registered in Japan and to set up provisional criteria for the new clinical entity IgG4-positive multiorgan lymphoproliferative syndrome (IgG4+MOLPS). The preliminary diagnostic criteria include raised serum levels of IgG4 (>135 mg/dl) and infiltration of IgG4+ plasma cells in the tissue (IgG4+/IgG+ plasma cells >50%) with fibrosis or sclerosis. The clinical features, laboratory data and pathologies of 64 patients with IgG4+MOLPS and 31 patients with typical SS were compared.
Results: The incidence of xerostomia, xerophthalmia and arthralgia, rheumatoid factor and antinuclear, antiSS-A/Ro and antiSS-B/La antibodies was significantly lower in patients with IgG4+MOLPS than in those with typical SS. Allergic rhinitis and autoimmune pancreatitis were significantly more frequent and total IgG, IgG2, IgG4 and IgE levels were significantly increased in IgG4+MOLPS. Histological specimens from patients with IgG4+MOLPS revealed marked IgG4+ plasma cell infiltration. Many patients with IgG4+MOLPS had lymphocytic follicle formation, but lymphoepithelial lesions were rare. Few IgG4+ cells were seen in the tissue of patients with typical SS. Thirty-eight patients with IgG4+MOLPS treated with glucocorticoids showed marked clinical improvement.
Conclusion: Despite similarities in the involved organs, there are considerable clinical and pathological differences between IgG4+MOLPS and SS. Based on the clinical features and good response to glucocorticoids, we propose a new clinical entity: IgG4+MOLPS.
Statistics from Altmetric.com
In 1888, Johann von Mikulicz-Radecki reported a man with symmetrical swelling of the lacrimal, submandibular and parotid glands of unknown aetiology.1 Histologically, the swollen glands showed massive mononuclear cell infiltration, and this condition is called Mikulicz’s disease (MD).2 3 Since Morgan et al reported in 1953 that MD was not a distinct clinical and pathological disease but merely one manifestation of Sjögren’s syndrome (SS),4 MD has attracted very little interest.
However, more than 20 cases of MD were reported between 1960 and 2006 in Japan, and differences between MD and SS have been investigated.5–10 Previous case reports indicated that MD may differ from SS in a number of respects:7–10 (1) MD occurs in both men and women whereas SS occurs mainly in women; (2) patients with MD show significant enlargement of the lacrimal and salivary glands but relatively mild xerostomia and xerophthalmia; (3) more complications such as autoimmune pancreatitis have been reported in MD; (4) raised levels of serum IgG4 and IgG4+ plasma cell infiltration in tissues were reported in patients with MD; (5) a better response to glucocorticoid therapy is achieved in patients with MD than in those with SS.
For analysis of IgG4-related disorders including MD, autoimmune pancreatitis and other conditions, we performed a retrospective analysis of data from patients with MD and similar cases from all over Japan. From these results, we propose a new clinical entity for these disorders with characteristic features: IgG4-positive multiorgan lymphoproliferative syndrome (IgG4+MOLPS).
METHODS
Case reports of MD involving two or three sets of swollen lacrimal, parotid or submandibular glands on physical examination and IgG4-related disorders have been collected from all over Japan since 2004. A total of 85 patients from 10 collaborating institutes were registered retrospectively. The diagnosis of IgG4+MOLPS was defined as both raised serum IgG4 levels (>135 mg/dl)11 and histopathological features including lymphocyte and IgG4+ plasma cell infiltration (IgG4+ plasma cells/IgG+ plasma cells >50%)12 13 with typical fibrosis or sclerosis in the tissue. Sixty-four of these 85 cases were diagnosed as IgG4+MOLPS. Of these 64 patients with IgG4+MOLPS (mean age 57.0 years; median age 60.0 years; range 17–80), 33 were women (mean age 54.7 years; median age 56.0 years; range 17–77) and 31 were men (mean age 59.4 years; median age 62.0 years; range 23–83).
These were compared with 31 patients with patients with typical primary SS (male:female ratio 2:29; mean age 52.0 years; median age 49.0 years; range 34–76). Typical patients with SS fulfilled both Japanese14 and European15 SS criteria and were positive for both anti-SS-A/Ro and anti-SS-B/La antibodies.
Histopathological findings were examined by haematoxylin and eosin staining and immunohistochemical staining using anti-CD3 antibody (rabbit polyclonal anti-human CD3 A0452; Dako, Glostrup, Denmark), anti-CD20 antibody (mouse monoclonal anti-human CD20 M0755; Dako), anti-CD38 antibody (mouse monoclonal anti-human CD38 NCL-CD38-290; Novocastra, Newcastle-upon-Tyne, UK), anti-IgG antibody (mouse anti-IgG antibody, M0828; Dako) and anti-IgG4 antibody (mouse anti-human IgG4 antibody MC011; The Binding Site, Birmingham, UK). Biopsy specimens of minor salivary glands from 22 typical patients with SS with marked lymphocytic infiltration were also examined by IgG4 immunostaining.
Laboratory data and the clinical response to treatment were investigated. The study was approved by the review board of Kanazawa Medical University and those of each collaborating institute. All data and samples from patients were collected with their informed consent.
Statistical analysis
The frequencies of symptoms, complications and laboratory data were compared between the groups. IgG4+MOLPS was seen in both men and women, while the majority of patients with typical SS were women. We therefore compared data of all patients with IgG4+MOLPS patients and only female patients with IgG4+MOLPS with those of typical SS patients. We also compared female vs male patients with IgG4+MOLPS. Comparisons between the two groups were performed using the χ2 or Fisher exact test with regard to the frequencies of symptoms of xerostomia, xerophthalmia and arthralgia, and complications of allergic rhinitis, bronchial asthma, autoimmune pancreatitis, interstitial nephritis and interstitial pneumonia, and the incidences of rheumatoid factor, antinuclear antibody, anti-SS-A/Ro antibody, anti-SS-B/La antibody and decreased CH50 (50% haemolytic unit of complement). Comparisons of immunoglobulin classes and subclasses (IgG, IgG1, IgG2, IgG3, IgG4, IgA, IgM and IgE) were performed using the Mann-Whitney U test. All analyses were performed using SPSS V.11 (SPSS, Chicago, Illinois, USA).
RESULTS
As shown in table 1, the numbers of patients with symptoms of xerostomia, xerophthalmia and arthralgia were significantly lower in all cases of IgG4+MOLPS than in typical cases of SS (32.8% vs 93.5%, 37.5% vs 87.1% and 15.6% vs 48.4%, respectively). Similar results were seen in the comparison of female patients with IgG4+AMOLPS compared with patients with typical SS. Allergic rhinitis and autoimmune pancreatitis were significantly more common in IgG4+MOLPS than in typical SS (40.6% vs 6.5%, 17.2% vs 0%, respectively). Interstitial pneumonitis was significantly rarer in all patients with IgG4+MOLPS than in patients with typical SS (9.4% vs 32.3%). We compared gender differences among IgG4+MOLPS cases and found that autoimmune pancreatitis was significantly more common in men than in women (32.3% vs 3%).
The incidences of rheumatoid factor, antinuclear antibody, anti-SS-A/Ro antibody and anti-SS-B/La antibody were significantly lower in patients with IgG4+MOLPS than in those with typical SS (table 2). The same tendency was seen when women with IgG4+MOLPS were compared with patients with typical SS. We compared immunoglobulin classes and subclasses and found that not only IgG4 but also total IgG, IgG2 and IgE levels were significantly higher in IgG4+MOLPS than in typical SS. In contrast, IgG1, IgG3, IgA and IgM levels were significantly lower in IgG4+MOLPS than in typical SS.
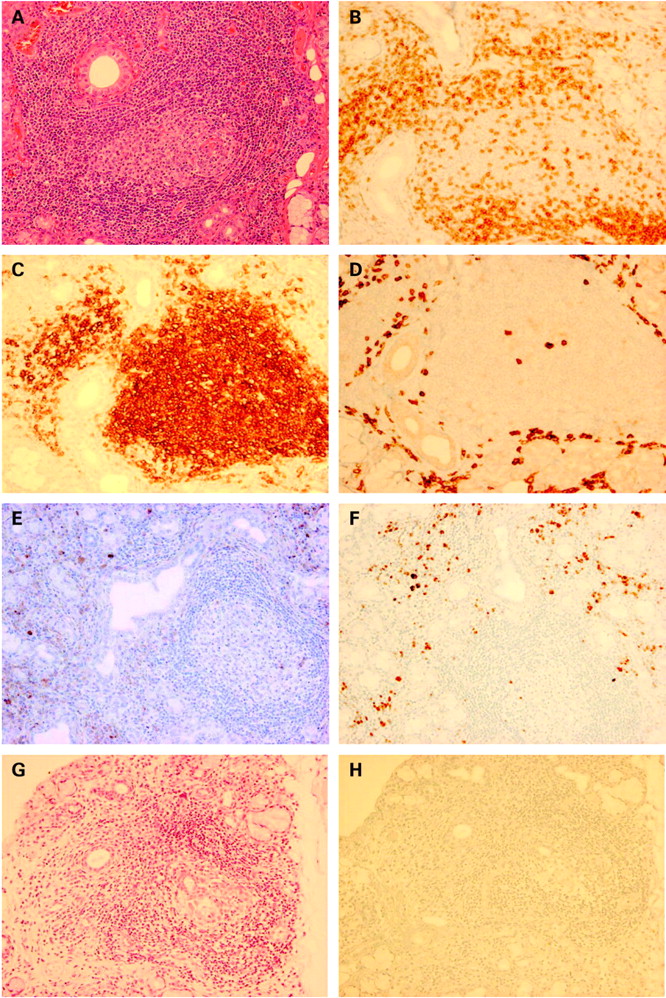
Patients with IgG4+MOLPS showed marked lymphocyte and IgG4+ plasma cell infiltration with fibrosis (sclerotic lesions). Furthermore, lymphocytic follicle formation was observed in many patients (fig 1). Lymphocytic infiltration into the ducts (formation of lymphoepithelial lesions) was rare, and many IgG4+ cells were scattered in the periphery of the follicles. In situ hybridisation of kappa and lambda indicated polyclonal B cell proliferation (see figure in online supplement). In contrast, few or no IgG4+ cells were seen in biopsy specimens of minor salivary glands from 22 patients with typical SS with severe lymphocytic infiltration.

Histopathological findings of labial minor salivary gland biopsy in (A–F) patients with IgG4+MOLPS/Mikulicz’s disease and (G, H) patients with Sjögren’s syndrome. (A, G) H&E staining; (B) CD3; (C) CD20; (D) CD38; (E) IgG; (F, H) IgG4 immunostaining. Massive lymphocyte and plasmacyte infiltration and lymphoid follicle formation were seen in IgG4+MOLPS. The ducts remained clearly without lymphocytic infiltration. CD20+ B cells remained in the follicle and CD3+ T cells were seen around the follicle. CD38+ plasma cells, IgG+ cells and IgG4+ plasma cells were scattered in the periphery of the follicle. The ratio of IgG4+ plasma cells/IgG+ plasma cells was >50%. On the other hand, there were few or no IgG4+ cells in typical SS, even in cases with severe lymphocytic infiltration.
Thirty-eight of the 64 patients with IgG4+MOLPS were treated with glucocorticoids. The starting dose of prednisolone was 10–30 mg/day for the majority of patients (n = 25), and higher doses of 40–60 mg/day for those patients suffering severe complications (n = 13) such as pancreatitis, interstitial nephritis, interstitial pneumonitis or hydronephrosis due to retroperitoneal fibrosis. Twenty-six patients were followed up without glucocorticoid treatment because their symptoms were mild or they refused glucocorticoid treatment. Glucocorticoid treatment markedly improved clinical signs and symptoms such as gland swelling (fig 2), but recurrence was seen in some cases (n = 15) when the glucocorticoid was tapered early or discontinued. A maintenance dose of 2.5–10 mg/day prednisolone was then used in most patients (37/38 patients).

Effect of glucocorticoid therapy on swollen lacrimal glands. (A–D) Photographs of face and eyes. (E–H) MRCT T1 imaging of another patient. (E, G) Frontal cross-section. (F, H) Coronal cross-section. (A, B, E, F) Before treatment, markedly swollen lacrimal glands were seen. (C, D, G, H) After glucocorticoid treatment, lacrimal swelling was reduced.
DISCUSSION
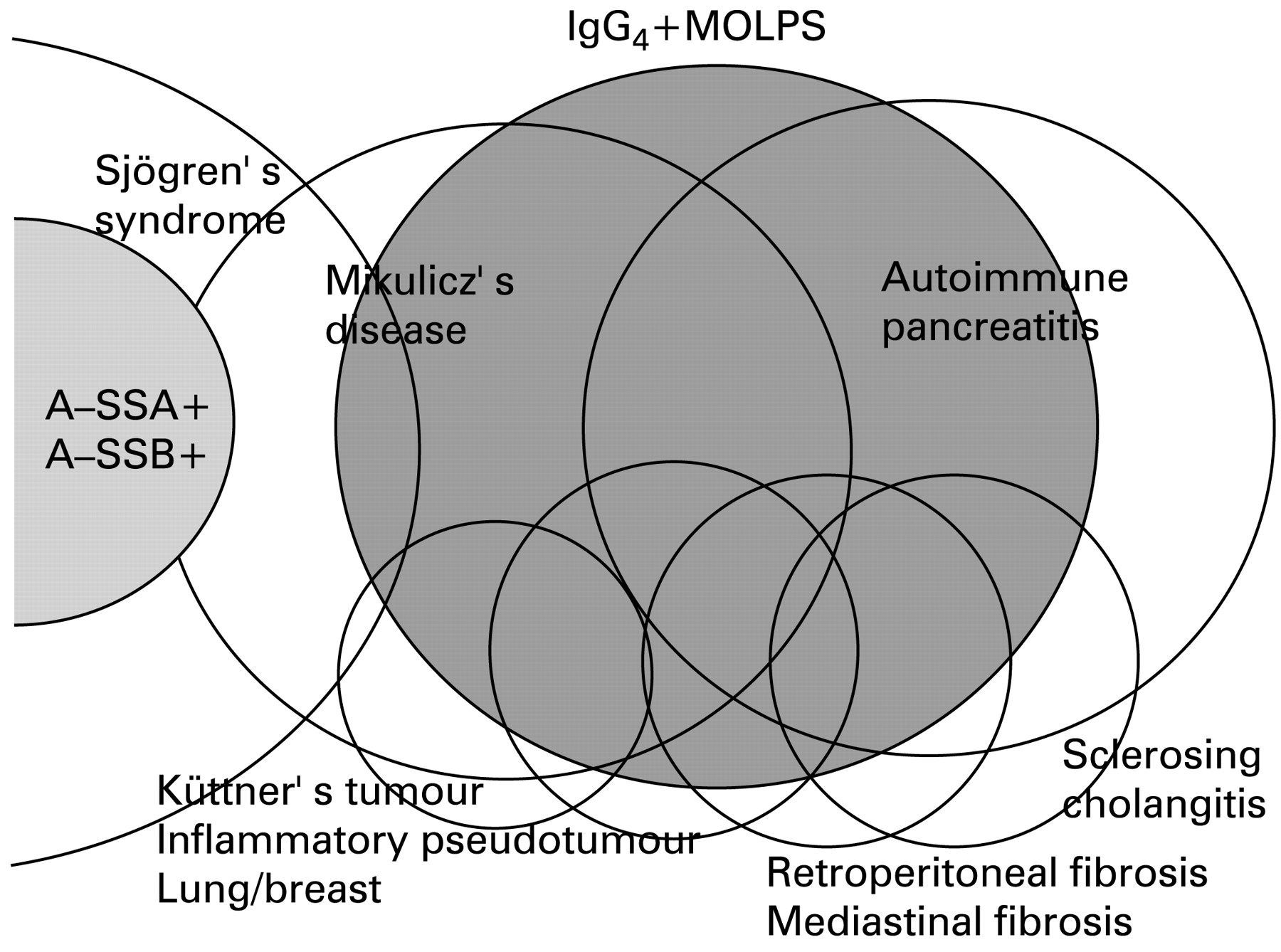
We propose a new clinical entity, IgG4+MOLPS, a syndrome characterised by hyper-IgG4 gammaglobulinaemia and IgG4+ plasma cell infiltration in the tissue (lymphoproliferative disorder). IgG4+MOLPS includes MD, autoimmune pancreatitis,11 16–21 sclerosing cholangitis,18 Küttner’s tumour,15 inflammatory pseudotumour of the lung,19 liver18 and breast,16 22 retroperitoneal and mediastinal fibrosis,20 interstitial nephritis,8 23 autoimmune hypophysitis9 and many other inflammatory conditions in multiple organs (fig 3). The distribution of involved organs in IgG4+MOLPS is similar to that in SS, but there are obvious differences in clinical and pathological features between these classifications. SS is also a very broad-spectrum syndrome, as both anti-SS-A/Ro antibody-positive and anti-SS-B/La antibody-positive SS patients are thought to be typical SS. We compared IgG4+MOLPS with 31 cases of typical SS.

{kind=link}
{kind=link}
{kind=link}
Spectrum of IgG4+MOLPS. IgG4+MOLPS included Mikulicz’s disease, autoimmune pancreatitis and many other inflammatory conditions. A-SSA, anti-SS-A/Ro antibody; A-SSB, anti-SS-B/La antibody.
The age range of patients with IgG4+MOLPS was broad in our series, ranging from 17 to 80 years with mean and median ages similar to those of SS. On the other hand, the gender distribution was quite different. Male patients with SS are very rare (2/31), but almost half (31/64) of the patients with IgG4+MOLPS were men. These results suggest that the differential diagnosis of IgG4+MOLPS should be re-examined in men with SS, even if they meet the SS criteria.
Although swollen glands are usually correlated with xerostomia and xerophthalmia in patients with SS, the incidence of xerostomia and xerophthalmia was significantly lower in IgG4+MOLPS, even in cases where the lacrimal, parotid or submandibular glands were swollen. Histopathological examination showed that lymphocytic infiltration in the ducts and formation of lymphoepithelial lesions are rare in IgG4+MOLPS, even in cases showing severe lymphocyte and plasma cell expansion. This may explain the marked swelling of the glands without severe dryness in IgG4+MOLPS. The decreased numbers of apoptotic cells and abnormal expression and function of Fas ligand in MD tissue5 6 are compatible with this observation.
Allergic rhinitis and bronchial asthma were more common in IgG4+MOLPS than in typical SS. This tendency towards an increased incidence of these allergic conditions may be related to the observation that IgG4 and IgE levels were significantly higher in IgG4+MOLPS than in SS. With regard to gender differences, autoimmune pancreatitis and interstitial nephritis were more common in men with IgG4+MOLPS than in women. Thus, the clinical picture was more severe in men with IgG4+MOLPS.
The incidence of rheumatoid factor, antinuclear antibody, anti-SSA/Ro antibody and anti-SSB/La antibody was significantly lower in IgG4+MOLPS than in SS. IgG4+MOLPS should therefore be suspected in patients with symptoms of SS but without autoantibodies, and IgG subclasses should be examined in such patients. Total IgG, IgG2, IgG4 and IgE were significantly higher and IgG1, IgG3, IgA and IgM were significantly lower in patients with IgG4+MOLPS than in patients with typical SS. The amount of immunoglobulin protein differed markedly between IgG4+MOLPS and typical SS and, thus, immunoglobulin gene usage, rearrangement pattern or regulation by T cells may be different in the two groups. The gene fragments Cγ2, Cγ4 and Cϵ—which code IgG2, IgG4 and IgE, respectively—line up side by side and therefore gene linkage may explain the observed association.
Histopathological differences are important to differentiate IgG4+MOLPS from SS. IgG4+ plasma cell infiltration in tissue was seen in patients with IgG4+MOLPS but not in those with SS. Expansion of IgG4+ plasma cells with fibrosis or sclerosis is an important histopathological finding in IgG4+MOLPS which is not usually seen in SS. Furthermore, lymphocytic follicle formation is commonly observed in IgG4+MOLPS but lymphocytic infiltration in the ducts (formation of lymphoepithelial lesions) is rare. In cases in which differential diagnosis is difficult, serum IgG subclasses and IgG4+/IgG+ immunostaining of tissue should be examined.
A good response to glucocorticoid therapy is usually seen in IgG4+MOLPS, and this is the most important reason for separating IgG4+MOLPS from SS. It will be necessary to develop guidelines for standard glucocorticoid therapy (indications, doses and tapering) based on the results of a larger study.
The differential diagnosis of IgG4+MOLPS from multicentric Castleman’s disease (MCD) or idiopathic plasmacytic lymphadenopathy (IPL) is important. Hyper-IgG4 gammaglobulinaemia and IgG4 plasma cell infiltration with fibrosis and sclerosis of tissue may be present in these conditions,24 and therefore discrimination may sometimes be difficult based on histological findings. On serological analysis, increased levels of interleukin-6 are seen in MCD and IPL but not in IgG4+MOLPS. IgG4+MOLPS shows a good response to glucocorticoid therapy while MCD and IPL do not. Measurement of the serum interleukin-6 level in such cases is therefore necessary for differential diagnosis and to determine whether glucocorticoids should be used. Patients with raised interleukin-6 levels should be diagnosed as having MCD or IPL, and not IgG4+MOLPS. An IgG4+/IgG+ cell ratio of >50% is usually not seen in MCD or IPL.
In Wegener’s granulomatosis, hyper-IgG4 gammaglobulinaemia and IgG4 plasma cell infiltration of tissue may be present.25 However, it is not difficult to discriminate between Wegener’s granulomatosis and IgG4+MOLPS based on the histological differences between these two conditions. IgG4+ plasma cells are also seen in other inflammatory or neoplastic conditions, and it is necessary to determine the IgG4+/IgG+ cell ratio to make a definite diagnosis of IgG4+MOLPS.
In conclusion, we propose a new clinical entity, IgG4+MOLPS, which has a similar distribution of involved organs to SS but has clinical (symptoms, complications, immunological data, including autoantibodies and immunoglobulin classes/subclasses) and histological (sclerosis or fibrosis and percentage of IgG4+ cells) differences. Owing to the good response to glucocorticoid treatment, IgG4+MOLPS should be excluded from the SS criteria. However, these conditions are still rare, and the aetiology and mechanism of development of IgG4+ cells are still unknown. It is therefore necessary to collect and analyse more of data from patients worldwide.
Acknowledgments
We thank the members of the MOLPS/Mikulicz’s Disease Society of Japan as follows: T Itho (Nagaoka Red-Cross Hospital); S Ideta (Keio University); Y Sasaki (Tokyo Dental University); K Fujibayashi (Dokkyo Medical University); Y Yamanishi (Hiroshima Citizen Hospital); A Kawakami, H Wada (Nagasaki University); M Uchida (Khochi University); K Nagasawa (Saga University); N Ogawa, K Shimoyama (Hamamatus Medical University); Y Hirose, H Minato, E Kinoshita, H Karasawa, M Miki, H Iwao, A Nakajima, Z-X Jin, C-R Huang, X-P Tong, Y Sun, K Fujimoto (Kanazawa Medical University). This work was supported by grants from the Japanese Ministry of Education, Culture, Sports, Science and Technology (13557160, 15024236, 15390313, 13877075, to Umehara and 17591060 to Masaki), Uehara Memorial Foundation (to Umehara) and Kanazawa Medical University Research Foundation (C2006-1 to Umehara and S2004-16, S2007-5 to Masaki).
REFERENCES
Supplementary materials
Web Only Data 68:8;1310-1315
Files in this Data Supplement:
{kind=link}
Footnotes
Competing interests: None.
Patient consent: Obtained.
Ethics approval: The study was approved by the review board of Kanazawa Medical University and those of each collaborating institute. All data and samples from patients were collected with their informed consent.
▸ Additional data are published online only at http://ard.bmj.com/content/vol68/issue8