Article Text

Abstract
Background and objectives Efficacy of adalimumab for ankylosing spondylitis (AS) has been established for Western populations but not in the Chinese population. This study is the first to evaluate the efficacy and safety of adalimumab in Chinese patients with AS.
Methods Chinese adults with active AS who had an inadequate response or were intolerant to ≥1 non-steroidal anti-inflammatory drugs were randomised to adalimumab 40 mg (N=229) or matching placebo (N=115) subcutaneously every other week (EOW) for 12 weeks, followed by a 12-week open-label adalimumab 40 mg EOW phase. The primary efficacy endpoint was the percentage of patients meeting the Assessment in Spondyloarthritis International Society (ASAS20) response criteria at week 12. The recently developed AS Disease Activity Score (ASDAS), as well as efficacy measures of spinal mobility, disease activity, physical function and quality of life were evaluated.
Results At week 12, adalimumab treatment resulted in a significantly greater percentage of ASAS20 responders than placebo (67.2% versus 30.4%, respectively; p<0.001). Differences in ASAS20 were observed as early as week 2 (42.8% vs 6.1%, respectively; p<0.001). The percentages of patients achieving ASAS40, ASAS 5/6 and ASDAS inactive disease were significantly greater with adalimumab than placebo at week 12 (all p<0.001). Tuberculosis was reported in one patient. No cases of malignancy, lymphoma, demyelinating disease or lupus-like syndrome were reported during the study.
Conclusions Adalimumab significantly reduced the signs and symptoms, improved physical function and quality of life of Chinese patients with active AS, and was generally safe and well tolerated in this population.
- Anti-TNF
- Ankylosing Spondylitis
- Disease Activity
- Treatment
Statistics from Altmetric.com
Introduction
Ankylosing spondylitis (AS), a disease characterised by inflammation of the axial skeleton, peripheral joints and entheses, can cause considerable disability and pain, similar to that caused by rheumatoid arthritis.1 The prevalence of AS in the Chinese population is estimated to be between 0.1% and 0.5%.2 In general, the clinical features and association with human leucocyte antigen B-27 (HLA-B27) of AS in Asian patients are similar to the rest of the world.3 HLA-B27 is strongly associated with AS and approximately 95% of Asian patients with AS are HLA-B27 positive, whereas 78–92% of patients in clinical studies of AS in Western populations were HLA-B27 positive.4–11 On the other hand, the association of interleukin-23 receptor (IL-23R) polymorphism with AS seen in Caucasian populations is not observed in Chinese Han populations.3 Prevalence of HLA-B27 varies with ethnicity and is approximately 8% among the general population in China.
Systemic corticosteroids and traditional disease modifying anti-rheumatic drugs (DMARDs) such as methotrexate and sulfasalazine have not been shown to be effective for the treatment of AS.12 ,13 Current Assessment in Spondyloarthritis International Society (ASAS)/European League Against Rheumatism recommendations are to treat AS patients failing conventional treatments (non-steroidal anti-inflammatory drugs (NSAIDs) and exercise) with anti-tumour necrosis factor (TNF) therapy.13 The efficacy and safety of several TNF-α inhibitors, including adalimumab,4 ,7 ,14 ,15 etanercept,10 ,16 infliximab8 and golimumab9 have been demonstrated in patients with AS in the USA and Europe. The efficacy of etanercept has also been demonstrated in Chinese patients with AS,17 ,18 with responses to treatment that are consistent with those observed in Western populations.10 ,11
The efficacy and safety of adalimumab for the treatment of rheumatoid arthritis has been demonstrated in a Chinese population19 and was consistent with results in Western populations.20 ,21 However, the efficacy and safety of adalimumab in Chinese patients with AS have not been previously reported. The objective of this study was to evaluate the safety and efficacy of adalimumab versus placebo in Chinese patients with AS who had an inadequate response or were intolerant to ≥1 NSAID.
Methods
Patients
Eligible patients were between 18 and 65 years of age, fulfilled modified New York Criteria for AS, had active disease (as defined by ≥2 of the following: Bath AS Disease Activity Index (BASDAI) ≥4 cm; total back pain on a visual analogue scale (VAS) ≥4 cm; and ≥1 hour of morning stiffness), and had an inadequate response or were intolerant to ≥1 NSAID. Patients with latent tuberculosis (TB) based on results of a positive purified protein derivative (PPD) test and chest radiograph had either completed or were receiving anti-TB therapy; patients with active, untreated TB were excluded from the study.
Patients were excluded from the study if they had total spinal ankylosis (bamboo spine); unstable extra-articular manifestations (eg, psoriasis, uveitis, inflammatory bowel disease); surgery involving the spine or joints within the previous 2 months; intra-articular or spinal/paraspinal corticosteroid injections within the previous 28 days; positive serology for HIV antibody, hepatitis B surface antibody or hepatitis C virus antibody; recent infection requiring anti-infectives; listeriosis; histoplasmosis; immunodeficiency syndrome; or chronic recurring infections. Patients with moderate to severe congestive heart failure, recent cerebrovascular accident, central nervous system demyelinating disease, or history of malignancy (except for successfully treated non-metastatic non-melanoma skin cancer or localised cervical carcinoma in situ) were also excluded.
Prior exposure to TNF-α inhibitors, natalizumab or efalizumab at any time, or use of traditional Chinese medicines within 28 days of baseline was not allowed. Concomitant use of methotrexate (≤25 mg/week), sulfasalazine (≤3 g/day), prednisone (≤10 mg/day), NSAIDs and/or analgesics was allowed but dose adjustments, induction and/or discontinuation of these therapies were only permitted during the open-label period. Other pharmacological therapies for AS except for those listed above could not be initiated at any time during the study.
Study design
This was a placebo-controlled, double-blind, randomised, phase III trial conducted between January 2010 and February 2011 at nine study sites in the People's Republic of China (NCT01114880). Following a screening period of up to 4 weeks, participants were centrally randomised using an interactive voice response or web-based system in a 2:1 ratio to receive adalimumab 40 mg or matching placebo subcutaneously every other week (EOW) during a 12-week double-blind phase. Study overseers, investigators, study site personnel and patients remained blinded to treatment during this phase, which was followed by a 12-week open-label phase, during which all patients received open-label adalimumab 40 mg EOW. A final follow-up visit for safety assessment was conducted 70 days after the last dose of study drug. All patients provided written informed consent. The study protocol was approved by the appropriate ethics committee and conducted in accordance with the International Conference on Harmonisation guidelines and the ethical principles of the Declaration of Helsinki.
Study endpoints
Efficacy and safety assessments were performed at weeks 0, 2 and 4 and every 4 weeks thereafter through the end of the open-label treatment phase. The primary efficacy endpoint was the percentage of patients achieving the ASAS20 response criteria at week 12, defined as an improvement of ≥20% and absolute improvement of ≥10 units from baseline in at least three of the following four domains: patient's global assessment of disease activity (PTGA; 0–100 mm VAS), total back pain (0–100 mm VAS), Bath AS Functional Index (BASFI), and inflammation/morning stiffness (mean questions 5 and 6 of the BASDAI), with no deterioration, defined as a change for the worse of ≥20% and a net worsening of ≥10 units, in the potential remaining domain. Additional endpoints assessed at weeks 12 and 24 were the percentage of patients achieving the following outcome measures: ASAS40, defined as improvement of ≥40% and absolute improvement of ≥20 units from baseline in at least three of the four domains listed for the ASAS20 with no deterioration (net worsening of >0 units) in the potential remaining domain; ASAS5/6, defined as 20% improvement in five of the following six domains: BASFI, total back pain, PTGA, inflammation/morning stiffness, spinal mobility (lateral lumbar flexion from Bath AS Metrology Index (BASMI)), and high-sensitivity C-reactive protein (hs-CRP); percentage of patients achieving ASAS partial remission, defined as a value of <20 in each of the four domains listed for ASAS20; and the percentage of patients achieving at least 50% improvement in the BASDAI score (BASDAI50).
The AS Disease Activity Score (ASDAS) is a recently developed composite measure incorporating questions 2 (neck, back or hip pain), 3 (joint pain/swelling) and 6 (duration of morning stiffness) of the BASDAI, the PTGA and hs-CRP.22 Using the ASDAS, patients were categorised into the following disease state categories: inactive (ASDAS score <1.3), moderate (≥1.3 to <2.1), high (≥2.1 to ≤3.5) and very high (>3.5). Clinically important improvement was defined as a decrease from baseline in the ASDAS of ≥1.1, and major improvement was defined as a decrease from baseline in the ASDAS of ≥2.0.
Additional efficacy endpoints assessed disease activity, pain and spinal mobility by measuring changes from baseline in PTGA (VAS), total back pain (VAS), inflammation/morning stiffness, BASDAI, physician's global assessment of disease activity (VAS), nocturnal pain (VAS), patient's global assessment of pain (VAS), tender joint count, swollen joint count, Maastricht AS Enthesitis Score (MASES), BASMI-linear and chest expansion. All measures recorded on a VAS were reported on a 0–10 cm scale. Health-related quality of life, physical function and work productivity measures included the Health Assessment Questionnaire modified for spondyloarthropathies (HAQ-S), 36-Item Short-Form Health Survey, V.2 (SF-36v2), BASFI, Bath AS Patient Global Index (BAS-G) and Work Productivity and Activity Impairment-Specific Health Problem Questionnaire (WPAI-SHP).
Safety assessments
Safety evaluations were conducted at every study visit and included adverse event (AE) monitoring and assessments of clinical laboratory and vital signs.
Statistical analysis
Assuming an expected ASAS20 response rate of 30% in the placebo group and 50% in the adalimumab group, the planned total study sample size of 330 patients was estimated to provide 94% statistical power to detect a difference between the two treatment groups.
The primary analysis of the ASAS20 response rate at week 12 in the adalimumab group versus the placebo group was performed using the two-sided Pearson χ2 test with α=0.05. Analysis of the primary endpoint was conducted for various subgroups to assess the impact of baseline variables on treatment response. A logistic model with treatment and a prespecified subgroup in the model was performed to assess the treatment by subgroup interaction. If the treatment by subgroup interaction was significant (p≤0.10), then treatment effect was assessed for components of the subgroup. For the primary endpoint and other categorical variables, a non-responder imputation approach was done at week 12 for missing data. Patients without data at week 12 were treated as non-responders. For continuous variables at week 12, missing data were imputed using the last observation carried forward. Differences from baseline between adalimumab and placebo groups were compared using an analysis of covariance method, adjusting for the baseline score. Open-label extension data at week 24 were summarised descriptively. All efficacy variables were analysed for the intent-to-treat (ITT) population, defined as all randomised patients who received ≥1 double-blind dose of study drug. The safety population consisted of all patients who received ≥1 dose of study drug. Data were analysed using SAS V.9.1 (SAS Institute, Inc, Cary, NC).
After translating the BASDAI from English to Chinese a printing error occurred. The incorrect version of the BASDAI instrument, which was missing the 0.5-h marker and had a different length for the 1-h mark for the VAS line of question 6 (duration of morning stiffness), was used before 26 April 2010. Patients who were assessed using this incorrect version (n=166) continued to use this version throughout the study, whereas patients who were screened on 26 April 2010 or later used the correct version of the BASDAI (n=178). The results presented for the ITT analysis have no adjustment for the incorrect version; however, additional analyses using an algorithm to convert scores from the incorrect BASDAI to a corrected score showed similar results to the ITT analysis. In addition, results from patients in the ITT population who used the correct version of the BASDAI were used in a sensitivity analysis of the ASAS20 responders to assess the impact of using the incorrect version of the BASDAI. Results were similar to the ITT analysis (data not shown).
Results
Patients and baseline characteristics
A total of 344 patients (115 randomised to placebo and 229 to adalimumab) were enrolled and received at least one double-blind dose of study drug (ITT set). There were 12 patients who prematurely discontinued from the study, and 332 patients completed the study (figure 1). No significant differences in patient baseline demographics and disease characteristics (table 1) were observed between groups, except for a significant difference in the ASDAS disease state categories (p=0.040), for which a higher proportion of patients in the adalimumab group were classified in the very high category versus the placebo group (65.5% vs 60.0%, respectively), and a smaller proportion of patients in the adalimumab group were classified in the moderate category versus the placebo group (2.6% vs 8.7%, respectively).
Patient demographics and baseline clinical characteristics

Patient disposition. AE, adverse event; EOW, every other week.
Efficacy assessments
For the primary endpoint, a significantly greater percentage of patients in the adalimumab group achieved an ASAS20 response at week 12, compared to the placebo group (67.2% vs 30.4%; p<0.001; figure 2). Furthermore, the ASAS20 rates were significantly different between the two treatment groups as early as week 2. Of the 75 (32.8%) adalimumab ASAS20 non-responders at week 12, 36 became responders at week 24. In the open-label period, the response rate after 4 weeks of treatment for placebo patients who were switched to open-label adalimumab (63.7%) was similar to the response observed in the original adalimumab group (59.0%) after 4 weeks of treatment in the double-blind period (figure 2). Subgroup analyses of the ASAS20 response at week 12 (non-responder imputation), based on a logistic regression model, showed that adalimumab was effective in many patient subgroups regardless of gender, age, weight, baseline hs-CRP, HLA-B27 status, disease duration or concomitant DMARD or NSAID use (see online supplementary table S1).

Percentage of ASAS20 responders over time. ASAS, Assessment in Spondyloarthritis International Society.*p<0.001 for difference between treatment groups; non-responder imputation. †Placebo, N=115; continuous adalimumab, N=229. ‡Open-label (OL) adalimumab, N=113; continuous adalimumab, N=224.
At week 12, efficacy of adalimumab treatment was demonstrated versus placebo for several secondary efficacy variables, excluding chest expansion (table 2, figure 3).
Mean change from baseline in efficacy variables
{kind=link}
{kind=link}
{kind=link}
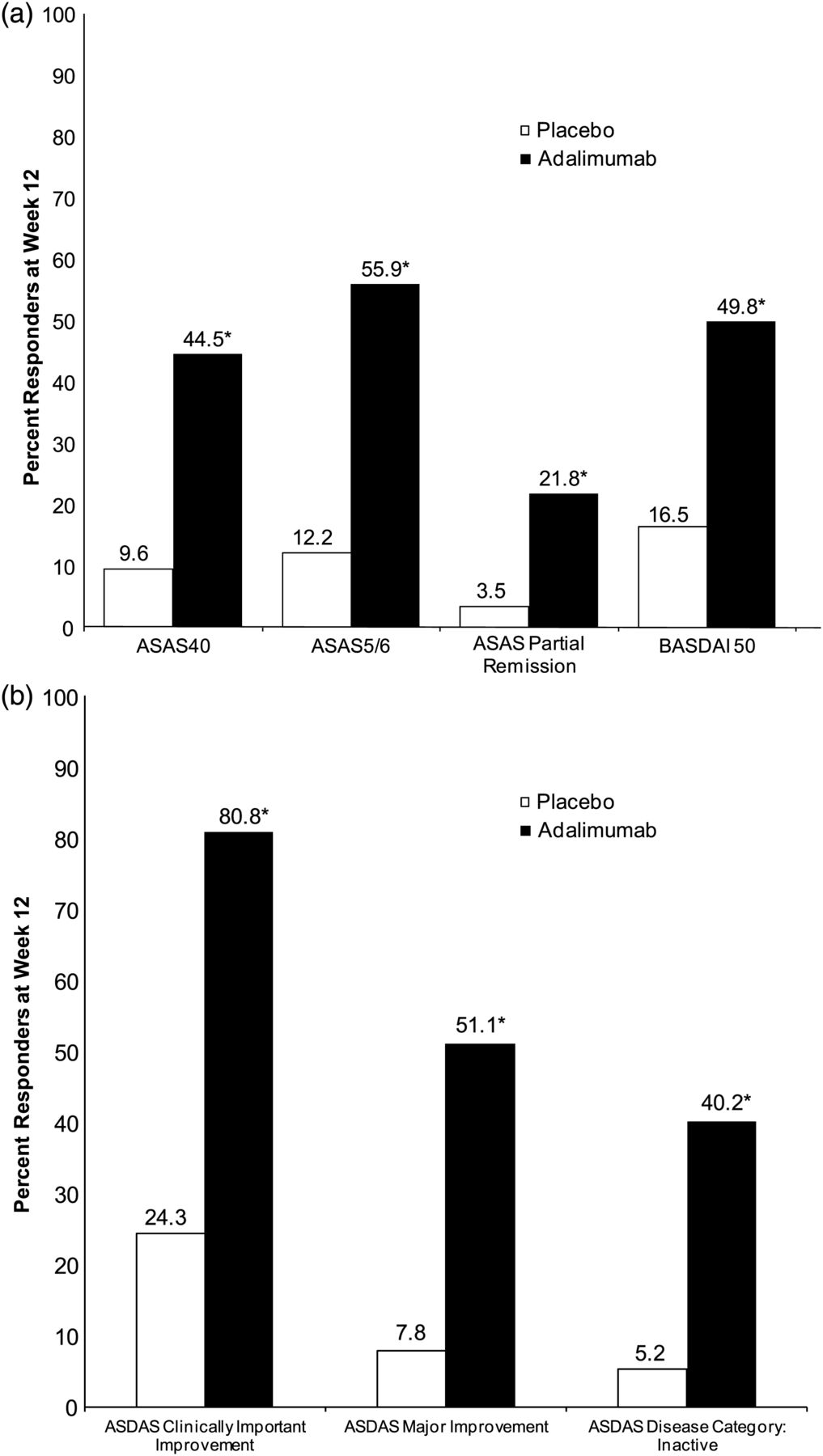
Percentage responders at week 12 for (A) ASAS and BASDAI responses and (B) ASDAS outcome measures. ASAS, Assessment in Spondyloarthritis International Society; ASDAS, Ankylosing Spondylitis Disease Activity Score; BASDAI, Bath Ankylosing Spondylitis Disease Activity Index. *p<0.001; non-responder imputation.
Adalimumab treatment was associated with significantly greater response rates than placebo for ASAS40, ASAS5/6, ASAS partial remission and BASDAI50 (figure 3A). Also at week 12, there were greater percentages of patients in ASDAS inactive disease (figure 3B) and moderate (28.8% vs 17.4%; p=0.021) categories and lower percentages of patients in the very high ASDAS disease category (3.5% vs 40.0%; p<0.001) with adalimumab versus placebo. Additionally, adalimumab-treated patients reported significant improvements in quality-of-life measures (HAQ-S and SF-36), and in activity impairment and overall work impairment (WPAI-SHP) compared with placebo at week 12 (table 2). Response rates for the secondary endpoints were similar between the two treatment groups by week 12 of the open-label period (week 24 of the study), regardless of the original randomised treatment received during the double blind period (table 2).
Safety assessments
During the double-blind period of the study, a numerically greater proportion of patients in the adalimumab group had a treatment-emergent AE (TEAE) compared with patients in the placebo group (35.4% and 22.6%, respectively; table 3). All AEs reported during the double-blind period of the study were considered mild or moderate in severity by the investigator. Serious AEs were reported by one patient in the placebo group (fibroadenoma of breast and intraductal papilloma of breast) and one patient in the adalimumab treatment group (pelvic inflammatory disease). Four patients in the adalimumab group discontinued during the double-blind period due to AEs, consisting of increased alanine aminotransferase (ALT) and increased aspartate aminotransferase (AST) in one patient and increased levels of ALT, AST, blood alkaline phosphatase and blood triglycerides in another patient, both of whom had a history of fatty liver disease; anaemia, thrombocytopenia and pulmonary fibrosis in one patient; and pelvic inflammatory disease in one patient. Of these four events, only the case of pelvic inflammatory disease was considered serious by the investigator.
Summary of treatment-emergent adverse events
An imbalance in hepatic-related AEs was observed between the placebo and adalimumab treatment groups during the double-blind period (table 3). All hepatic-related AEs reported in the adalimumab group were laboratory abnormalities in ALT, AST or alkaline phosphatase; most were considered mild by the investigator and did not result in patient discontinuation from the study.
During the open-label period of the study, four serious AEs were reported: concussion, contusion and skin laceration in one patient; elective abortion in one patient; peritoneal TB, pulmonary TB and TB pleurisy in one patient; and viral hepatitis in one patient. The patient with TB was a 36-year-old man with a negative PPD screening test and non-clinically significant increased lung markings on the screening chest x-ray who had contact with a patient with TB during the study. The patient with viral hepatitis was a 27-year-old man living with family members who were positive for hepatitis B. No cases of malignancy, lymphoma, opportunistic infections other than TB, demyelinating disease, congestive heart failure, diverticulitis, leucoencephalopathy or lupus-like syndromes were reported. No deaths occurred during the study.
Discussion
The results of this study show that adalimumab reduces the signs and symptoms of active AS in adult Chinese patients and provides additional evidence that adalimumab is effective in patients with AS. Efficacy of adalimumab was consistently demonstrated compared with placebo across multiple measures of disease activity, spinal mobility, physical function, quality of life and work productivity. Adalimumab was also associated with a rapid onset of action, with significant results seen after 2 weeks of treatment. Further, adalimumab was safe and generally well tolerated for up to 24 weeks of therapy in this patient population.
The Chinese AS population enrolled in this trial was similar to the Western population in the Adalimumab Trial Evaluating Long-Term Efficacy and Safety for Ankylosing Spondylitis (ATLAS) study in terms of baseline demographics and disease characteristics. However, the mean age of the Chinese study population was approximately 12 years younger than that of the Western study population; concomitant use of DMARDs was higher in the Chinese study population than in the Western population (60% vs 20% of patients); and fewer subjects reported a history of uveitis, psoriasis or inflammatory bowel disease in this study compared with subjects in the ATLAS trial.4 Efficacy results in the current study were consistent with those seen with adalimumab in Western patients with AS.4 ,15 In the ATLAS trial, ASAS20 was achieved at week 12 by 58% and 21% of patients on adalimumab or placebo, respectively, compared with 67.2% and 30.4% in this study.4 Recently, the ABILITY-1 trial also demonstrated the efficacy of adalimumab for patients with non-radiographic axial spondyloarthritis, with ASAS20 response rates at week 12 significantly higher in the adalimumab group compared with placebo (52% vs 31%).23 Clinical response to adalimumab as measured by ASAS40 was also similar in the current study, the ATLAS trial and the ABILITY-1 trial (44.5%, 39.9% and 36%, respectively).4 ,23
Adalimumab resulted in a significantly higher percentage of ASAS20 responders compared with placebo, and also resulted in a significantly higher percentage of patients achieving clinically important improvement and major improvement as determined by ASDAS. This study is the first large, double-blind, randomised controlled study to use the ASDAS as a predefined endpoint in either Western or Chinese populations. Compared with the ASAS20 and BASDAI, the ASDAS includes an objective measure of disease activity (hs-CRP), similar to the ASAS 5/6.22 ,24 As patients and physicians are known to often have differing perceptions of disease activity,25 an objective measure of inflammation thus increases the face validity of the ASDAS. The ASDAS was recently validated in a Chinese population with AS.26
The chronic course of AS can result in significant disability, poor quality of life and decreased work productivity.1 With AS generally beginning in early adulthood, often considered the most productive years of life, there may be considerable costs related to both health care and inability to perform paid work. Therefore, the improvements in physical function, quality of life, and work productivity measures demonstrated with adalimumab treatment may be particularly pertinent for the relatively young patient population in this study.
Few AEs of special interest for TNF inhibitors were observed in this study.27 ,28 With thorough screening for TB and hepatitis B, infections that are both endemic to China, prior to study enrolment only one case each of TB and hepatitis B were reported in patients with known exposure to infected individuals during study participation. This highlights the utility of screening patients for TB29 and hepatitis B before initiating treatment with TNF inhibitors.27
Liver enzyme elevation has been reported with adalimumab therapy.27 Elevations in ALT and AST were the most common TEAEs observed in patients exposed to adalimumab at any time during the study, most of which were assessed as mild. None were severe. The ALT elevations seen in this study were generally similar to those observed in the ATLAS study.4
A limitation of this study was the relatively short treatment and observation period (up to 24 weeks), resulting in a lack of information regarding long-term efficacy and safety for the Chinese AS population. Although ASDAS, BASFI and BASDAI have been previously validated in Chinese patients with AS,26 ,30 other outcome measures used in this study have not previously been validated in the Chinese population; however, all are accepted for use in the wider AS clinical trial population.
In conclusion, adalimumab was an effective treatment for reducing signs and symptoms, as well as improving physical function and quality of life of Chinese patients with active AS and inadequate response or intolerance to NSAID therapy. Adalimumab appeared to be safe and well tolerated in this population, with a safety profile similar to that seen in Western patient populations.
Acknowledgments
Medical writing support was provided by Margit Rezabek, DVM, PhD, at Complete Publication Solutions, LLC; this support was funded by AbbVie.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online table
Footnotes
-
Handling editor Tore K Kvien
-
Funding This study was sponsored by AbbVie.
-
Competing interests FH is a consultant and has served on speakers bureaus for AbbVie China. CB has served on a speakers bureau for Pfizer. JX has served on a speakers bureau for AbbVie. HW has served on speakers bureaus for AbbVie and Xian Janssen. GW has served on a speakers bureau for Xian Janssen. QS has served on a speakers bureau for Pfizer and has received payment for the development of educational presentations from Roche. All non-AbbVie authors declare study grant support was given by AbbVie to their institution for conducting the study. NA, JA and AP are employees of and shareholders in AbbVie.
-
Contributors All non-AbbVie authors contributed to the collection and interpretation of study data. NA contributed to the analysis and interpretation of data. JA and ALP contributed to the interpretation of the data. FH is the guarantor of the manuscript.
-
Ethics approval Multiple independent ethics committees.
-
Provenance and peer review Not commissioned; externally peer reviewed.