Article Text

Abstract
Objective To assess survival and identify predictors of survival in patients with systemic sclerosis-interstitial lung disease (SSc-ILD) who participated in the Scleroderma Lung Studies (SLS) I and II.
Methods SLS I randomised 158 patients with SSc-ILD to 1 year of oral cyclophosphamide (CYC) vs placebo. SLS II randomised 142 patients to 1 year of oral CYC followed by 1 year of placebo vs 2 years of mycophenolate mofetil. Counting process Cox proportional hazard modelling identified variables associated with long-term mortality in SLS I and II. Internal validation was performed using joint modelling.
Results After a median follow-up of 8 years, 42% of SLS I patients died, and when known the cause of death was most often attributable to SSc. There was no significant difference in the time to death between treatment arms in SLS I or II. Higher baseline skin score, older age, and a decline in the forced vital capacity (FVC) and the diffusing capacity for carbon monoxide (DLCO) over 2 years were independently associated with an increased risk of mortality in SLS I. The Cox model identified the same mortality predictor variables using the SLS II data.
Conclusion In addition to identifying traditional mortality risk factors in SSc (skin score, age), this study demonstrated that a decline in FVC and DLCO over 2 years was a better predictor of mortality than baseline FVC and DLCO. These findings suggest that short-term changes in surrogate measures of SSc-ILD progression may have important effects on long-term outcomes.
- systemic sclerosis
- interstitial lung disease
- treatment
- survival
- systemic sclerosis
- interstitial lung disease
- treatment
- survival
Statistics from Altmetric.com
- systemic sclerosis
- interstitial lung disease
- treatment
- survival
- systemic sclerosis
- interstitial lung disease
- treatment
- survival
Key messages
What is already known about this subject?
Observational studies have identified factors associated with an increased risk of mortality in systemic sclerosis (SSc), including age, extent of cutaneous sclerosis and severity of interstitial lung disease (ILD).
What does this study add?
In addition to identifying traditional risk factors for mortality, this study found that short-term progression of ILD was a better predictor of mortality than baseline severity of ILD.
Specifically, patients who experienced a decline in lung function and diffusing capacity over 2 years had a substantially increased risk of mortality even after adjusting for treatment arm assignment, as well as baseline disease severity.
How might this impact on clinical practice or future developments?
Patients with SSc-ILD who experience an early decline in lung function and have an increased risk of death may benefit from receiving a more aggressive treatment approach that could include escalation of immunosuppression, addition of antifibrotic therapy and/or evaluation for haematopoetic stem cell transplant.
SSc providers should closely monitor lung function when ILD is present to accurately identify declines in lung function and promptly intervene to improve patient outcomes.
Introduction
Interstitial lung disease (ILD) is the leading cause of death in systemic sclerosis (SSc),1 accounting for over one-third of SSc-related deaths in a multicentre observational study of over 5000 patients with SSc.2 In addition, ILD occurs in the majority of patients with SSc3 and is found in 79% of patients with SSc at autopsy.4
Immunosuppressant agents, such as mycophenolate motetil (MMF) and cyclophosphamide (CYC), are currently used to treat SSc-ILD.5–7 However, randomised controlled trials (RCTs) have demonstrated that while some patients experience improvement in lung function after treatment with MMF or CYC, other patients experience ILD progression despite treatment with immunosuppression.6 7 Moreover, not all patients with ILD develop symptoms or will have progressive disease that leads to death even in the absence of treatment.3 8
The present study sought to develop a mortality prediction model using data from two RCTs for SSc-ILD (Scleroderma Lung Study (SLS) I and II).6 7 Using data from RCTs (in contrast to an observational cohort) may minimise confounding due to factors that affect survival, such as timing of treatment initiation, access to healthcare, socioeconomic status as well as comorbid conditions. From these two RCTs with rigorous entry criteria, close monitoring of pulmonary function every 3 months for 2 years and standard treatment regimens, the hypothesis was that in this controlled setting of 300 participants with SSc and ILD new predictors of mortality could be discovered.
Methods
Study participants
All participants enrolled in SLS I6(NCT01762449; NCT00004563) and SLS II7 (NCT00883129) were eligible to participate in the SLS long-term follow-up study. SLS I and II included adult patients with SSc with evidence of ILD on high-resolution CT (HRCT) with a duration of disease ≤7 years from onset of the first non-Raynaud’s symptom of SSc. (Please see online supplementary appendix for complete eligibility criteria.) Only participants who provided informed consent were included in the present analyses.
Supplemental material
SLS I and II study design
In SLS I, 158 participants were randomised to receive either oral CYC or matching placebo for 1 year, followed by an additional year of observation off-treatment as previously published.6 In SLS II, 142 patients were randomised to receive either MMF for 2 years or oral CYC for 1 year, followed by an additional year placebo using a double-dummy design to maintain the blinding as reported.7
SLS I and II assessment measurements
Complete details of SLS I and II assessment measurements are in the online supplementary appendix. Forced vital capacity (FVC) (primary SLS I and II endpoint) and diffusing capacity for carbon monoxide (DLCO) (secondary SLS I and II endpoint) were measured every 3 months during the 24-month study period for both studies.6 7 HRCT thoracic imaging was obtained at baseline and 24 months in SLS II and at baseline and 12 months in SLS I. Quantitative imaging analysis (to quantify the extent of ILD) was performed as previously published and is described in the online supplementary appendix.32
Long-term follow up assessment
During both the SLS I and II study periods, when the statistical centre was informed of a participant’s death, clinical research associates were asked to collect source documentation to determine the cause of death. A mortality and morbidity committee adjudicated the causes of death to determine whether the cause was related to underlying SSc, medication or another cause based on expert consensus. Following the 24-month study periods, patients or designated surrogates were contacted to assess morbidity and mortality outcomes. (Please see online supplementary appendix for further details of the long-term follow-up assessment.)
Statistical analysis
Baseline characteristics
Summary statistics were generated for baseline characteristics from the two cohorts. Group comparisons were performed using two-sample t-tests and χ2 tests.
Primary outcome: survival
The primary outcome was survival. The Kaplan-Meier estimate was used to generate survival curves, and the log-rank test was used to compare survival between groups. If survival status was unknown, survival time was censored at the date when the participant was last known to be alive. Cox proportional hazard models were developed to evaluate the impact of covariates shown previously to be associated with survival, including treatment, baseline Modified Rodnan Skin Score (MRSS), age, sex, race, disease duration, type of SSc (limited or diffuse), serological subtype (Scl-70 antibody-positive, RNA polymerase III antibody-positive), % predicted values for FVC or DLCO, and the radiographic quantitative extent of ILD/fibrosis. Models for FVC and DLCO were first fit using the baseline measure as the covariate of interest, and then in separate models using the longitudinal assessments over 24 months as a time-varying covariate. Final models were validated by fitting joint models for longitudinal and survival data using the SAS macro, JMFit9 (see online supplementary appendix for details on variables’ definitions and selection and joint modelling methods).
All tests were two-sided. All analyses were performed using SAS V.9.4.
Results
Participant characteristics
The baseline characteristics of the participants in SLS I and SLS II were fairly similar (table 1). SLS II participants were slightly older and had shorter disease durations compared with SLS I participants. While the FVC%-predicted did not differ between the two cohorts, SLS II participants had slightly more restrictive ventilatory impairment, as reflected by lower total lung capacity, despite less diffusion impairment.
Baseline characteristics of SLS I and SLS II participants
Participant disposition
SLS I
Twelve years after the first patient was randomised in SLS I, 66 of 158 (42%) participants had died (CYC: 38; placebo 28). Among the 37 patients for whom the cause of death was known, 24 deaths (65%) were attributable to underlying SSc, of which 16 (CYC 8; placebo: 8) were due to respiratory failure (table 2). Two of the deaths (1 CYC, 1 placebo) due to ‘Respiratory Failure’ were not attributed to underlying SSc. Survival status could not be determined in 34 participants. The median follow-up time for all patients in SLS I was 8 years.
Long-term morbidity and mortality outcomes of SLS I and II participants
SLS II
Eight years after the first patient was randomised in SLS II, 30 of 142 (21%) participants had died (CYC: 16; MMF: 14). Among the 26 patients for whom the cause of death was known, 15 deaths (58%) were attributable to underlying SSc, of which 13 (CYC: 6; MMF: 7) resulted from respiratory failure (table 2). Survival status could not be determined in 12 participants. The median follow-up time for all patients in SLS II was 3.6 years.
CYC does not improve long-term survival compared with placebo in SLS I
During the 24-month study period of SLS I, six participants randomised to CYC and six participants randomised to placebo expired.10 During the 12-year long-term follow-up period, there was no significant difference in the time to death (p=0.335 by log-rank test; figure 1A) nor the time to death or organ failure (p=0. 539 by log-rank test; figure 1B) for patients randomised to CYC versus placebo in SLS I. Moreover, time to the development of organ failure did not differ between the two study arms (p=0.185 by log-rank test; online supplementary figure S1), nor did time to the development of malignancy (p=0.701 by log-rank test; online supplementary figure S2). Types/locations of malignancies in SLS I included the anus (n=1), colon (n=2), vulvar (n=1), prostate (n=1), sarcoma (n=1) and breast (n=1) within the CYC arm, and colon (n=1), oesophageal (n=1), lung (n=3) and Hodgkin’s lymphoma (n=1) within the placebo arm.

Time to death (A) and time to death or organ failure (B) from randomisation in SLS I. There was no significant difference in the time to death (p=0.335 by log-rank test; A) nor the time to death or organ failure (p=0. 539 by log-rank test; B) for patients randomised to CYC versus placebo in SLS I. The last known date they were known to be alive was used for the survival analysis. CYC, cyclophosphamide; SLS, Scleroderma Lung Studies.
There is no difference in long-term survival between patients randomised to MMF versus CYC in SLS II
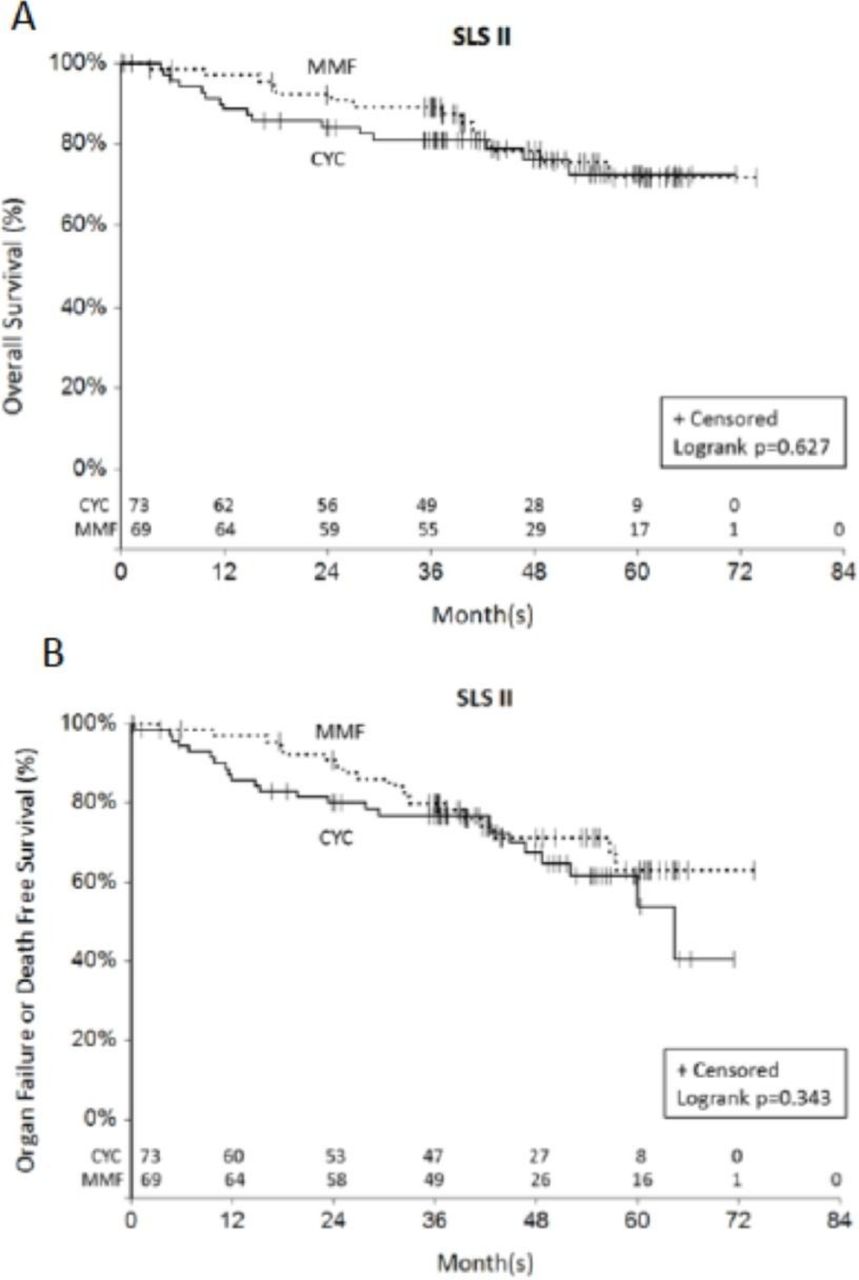
Over twice as many deaths occurred in the CYC arm (n=11) compared with the MMF arm (n=5) during the 24-month study period in SLS II (p=0.160 by log-rank test).7 During the 8-year long-term follow-up period, an additional five deaths occurred in the CYC arm compared with an additional nine deaths in the MMF arm. There was no significant difference in the time to death (p=0.627 by log-rank test; figure 2A) nor the time to death or organ failure (p=0.343 by log-rank test; figure 2B) for patients randomised to CYC versus MMF in SLS II. However, there appeared to be a separation in the survival curves favouring MMF within the first 2 years (figure 2A). There was no significant difference in the time to the development of organ failure between the two groups (p=0.692 by log-rank test; online supplementary figure S3). Two malignancies occurred during the follow-up period (MMF: n=1 thyroid cancer, n=1 papillary urothelial carcinoma; CYC: none).

{kind=link}
{kind=link}
Time to death (A) and time to death or organ failure (B) from randomisation in SLS II. There was no significant difference in the time to death (p=0.627 by log-rank test; (A)) nor the time to death or organ failure (p=0.343 by log-rank test; (B)) for patients randomised to CYC versus MMF in SLS II. The last known date they were known to be alive was used for the survival analysis. CYC, cyclophosphamide; MMF, mycophenolate mofetil; SLS, Scleroderma Lung Studies.
Longitudinal assessments of FVC and DLCO predict long-term survival in SLS I and II
SLS I: Cox proportional hazards models and joint models
The basic model developed from the SLS I cohort, as described in the online supplementary methods, consisted of the following covariates: treatment arm (CYC vs placebo), baseline extent of cutaneous sclerosis (MRSS), age at randomisation (years) and sex. Among these variables, increased age and MRSS were associated with increased mortality. The following variables were independently associated with mortality in the final models: (1) baseline FVC%-predicted; (2) longitudinal assessment of the FVC%-predicted measured as a time-varying covariate over 24 months; and (3) longitudinal assessment of the DLCO%-predicted measured as a time-varying covariate over 24 months (table 3). None of the quantitative lung fibrosis/ILD scores was associated with long-term survival when added to the base model. The Akaike information criterion (AIC),11–13 which estimates the quality of each model relative to each of the other models, was slightly lower (better) for the models that included the longitudinally measured FVC and DLCO parameters compared with those that included the baseline FVC and DLCO parameters (table 3). Thus, the final SLS I survival models demonstrated that decreased age, decreased extent of cutaneous sclerosis, as well as an improved course of the FVC%-predicted and the DLCO%-predicted over 24 months were associated with better survival outcomes (table 3). As stated in the online supplementary methods, we created separate models for the FVC and DLCO variables to avoid collinearity.
Final models for predicting death in SLS I
SLS I: joint model validation analysis
Using the same basic model as above (eg, MRSS, age), the longitudinal assessment of the FVC%-predicted was significantly associated with the outcome (table 3). When added to the basic model, the longitudinal assessment of the DLCO%-predicted was also significantly associated with the outcome (table 3). As noted above, the programming for the joint model does not allow for the inclusion of two longitudinally measured covariates simultaneously.
SLS I: exploratory analyses
In an exploratory analysis, we examined whether the change from baseline in the FVC%-predicted and DLCO%-predicted at 12 months predicted survival. When added to the basic model (eg, MRSS, age), none of the change scores was significantly associated with survival. In addition, we explored whether combined, categorical changes in the FVC%-predicted and DLCO%-predicted at 12 and 24 months predicted survival. None of the categorical declines at 12 months (eg, FVC decline ≥10%; FVC decline ≥15%; DLCO decline ≥15%; FVC decline ≥10% and DLCO decline ≥15%; FVC decline ≥10% or DLCO decline ≥15%) was significantly associated with long-term survival when added to the basic model; however, these individual categorical declines at 24 months were associated with long-term survival when added to the basic model (online supplementary table S1). Too few (patient (n=1) experienced an FVC decline 5%–9% and DLCO decline ≥15% to include this covariate in the model).
SLS II: Cox proportional hazards model
The basic model developed from the SLS II cohort consisted of the following covariates: treatment arm (CYC vs MMF), baseline extent of cutaneous sclerosis (MRSS), age at randomisation (years) and sex. Similar to SLS I, increased age and increased MRSS at baseline were associated with increased mortality in SLS II. Baseline FVC%-predicted and baseline DLCO%-predicted were not significantly associated with time to death when added to the basic model that comprised age and MRSS. However, the longitudinal assessment of the FVC%-predicted and the longitudinal assessment of the DLCO%-predicted were each associated with the outcome when added to the basic model (table 4). None of the quantitative lung fibrosis/ILD scores was associated with long-term survival when added to the base model. Therefore, the final SLS II survival models demonstrated that decreased age, decreased extent of cutaneous sclerosis, as well as an improved course of the FVC%-predicted and the DLCO%-predicted over 24 months were associated with better survival outcomes (table 4).
Final models for predicting death in SLS II
SLS II: joint model validation analysis
Using the same basic model as above (eg, MRSS, age), the longitudinal assessment of the FVC%-predicted was significantly associated with the outcome (table 4). When added to the basic model, the longitudinal assessment of the DLCO%-predicted was also significantly associated with the outcome (table 4).
SLS II: exploratory analyses
In an exploratory analysis, we examined whether the change from baseline in the FVC%-predicted and DLCO%-predicted at 12 months predicted survival. When added to the basic model (eg, MRSS, age), the change in FVC from baseline to 12 months predicted long-term survival (estimate 0.52 (CI 0.31 to 0.90); p<0.05), but not the change in DLCO from baseline to 12 months. In addition, we explored whether combined, categorical changes in the FVC%-predicted and DLCO%-predicted at 12 months predicted survival as described above. We found that an FVC decline ≥10% at 12 months and FVC decline ≥15% at 12 months were each associated with long-term survival when added to the base model with the covariates of MRSS and age (online supplementary table S2). Patients in SLS II had a decline in DLCO ≥15% at 12 months in SLS II; therefore, we were unable to analyse any of the the composite categorical decline variables that included the DLCO.
Use of potential disease-modifying agents
While no data were available regarding the use of potential disease-modifying agents beyond the 24-month study period in SLS I, in SLS II these data were collected and demonstrated that the majority of patients consumed MMF following the 24-month study period. (Please see online supplementary table S3 for a list of all potential disease-modifying agents used following cessation of study drug in SLS II.)
Discussion
The present study is the first study to examine mortality outcomes in patients who participated in two of the largest RCTs for SSc-ILD. The results presented herein demonstrate that treatment with 1 year of CYC compared with placebo does not improve long-term survival outcomes in patients with SSc-ILD. The findings also demonstrate that there is no difference in long-term survival between patients randomised to CYC versus MMF in SLS II.
Both SLS I and II demonstrated that treatment with immunosuppression led to short-term improvements in surrogate measures of SSc-ILD outcomes.6 7 However, the present findings suggest that short-term treatment with CYC and MMF may not improve long-term outcomes of patients with SSc-ILD. Where known, the majority of patients in both SLS I and II died of complications related to SSc, and respiratory failure due to end-stage lung disease was the leading cause of death. These findings are consistent with recent reports of mortality outcomes in observational cohorts.1 2
Relatively few malignancies occurred in either cohort. Furthermore, although studies have demonstrated an increase in haematological malignancies in SSc,14 only one case of lymphoma occurred in the placebo arm of SLS I. The paucity of malignancies observed in both cohorts may in part be due to the length of the follow-up period, especially in SLS II where the median follow-up was only 4 years, as well as the observation that respiratory failure was the leading cause of death.
These findings highlight a need to determine the appropriate duration of treatment for SSc-ILD. In SLS I, very few patients reported use of immunosuppression in year 2 of the study despite the accepted view that ILD progression generally occurs up to 5 years from disease onset in SSc. This may explain why there was no difference in long-term survival between the two study arms in SLS I. It is plausible that initiation of a maintenance therapy regimen after induction therapy may affect long-term survival outcomes. In SLS II, many patients continued on immunosuppression after the trial concluded (MMF was most common). The use of MMF in the CYC arm may in part explain why the trend for an MMF-related survival benefit observed in the first 2 years diminished in the subsequent follow-up years.
More research is needed to determine the appropriate length of treatment for immunosuppression in SSc-ILD. A prior small retrospective study demonstrated improvement/stability in the FVC after 18 months of azathioprine (AZA) maintenance therapy in patients who first completed a 6-month induction course of intravenous CYC for SSc-ILD.15 However, without a control arm, it is impossible to discern whether the observed improvement/stability in the FVC represents a true treatment response versus the natural course of ILD in SSc. An additional study demonstrated that patients who responded favourably to pulse CYC and were subsequently treated with AZA experienced a higher rate of improvement or stabilisation in lung function compared with patients who did not respond to pulse CYC.16
The present analysis also revealed significant predictors of long-term mortality in SSc-ILD. In line with prior observational studies,17–19 increased skin score and increased age were independently associated with increased mortality. In contrast to prior observational studies,18 20–22 however, male gender and African–American race were not associated with an increased risk of mortality. Regarding gender, the SLS I and II cohorts comprised predominantly of women; thus, these studies may be underpowered to detect true gender differences in long-term survival. In terms of race, our findings could potentially suggest that in the context of a clinical trial, in which all patients have equal access to healthcare and uniform follow-up, race does not play a substantial role in predicting long-term survival.
Consistent with prior observational studies,16 19 23 24 low baseline FVC was associated with an increased risk of mortality. However, the course of the FVC and the DLCO over 24 months appeared to be more robust predictors of long-term survival in both SLS I and II than the baseline measurements of these parameters when comparing the AIC for the models. The individual parameter estimates were similar for both the Cox model and the joint model that we used as a validation approach, suggesting that the relationship between survival and FVC (or DLCO) is not biased by non-ignorable missing data.
A recent single-centre observational cohort study of patients with SSc-ILD also found that pulmonary function trends at 1 and 2 years predicted intermediate to long-term mortality.25 This study demonstrated that 1-year categorical trends in the FVC and DLCO were the most accurate prognostic determinants of mortality, while at 2 years changes in gas transfer were the most important predictors of mortality.25 As this was an observational study, the authors could not adequately control for treatment effect and selection bias. We were able to replicate some, but not all of these findings in the SLS I or II cohorts. Taken together, the findings of the present study provide further evidence that trends in pulmonary function may offer more prognostic information than baseline pulmonary function measurements. This may in part be due to the fact that substantial variability exists in a single FVC and DLCO measurement. Repeated measurements of the FVC/DLCO may yield more clinically meaningful information regarding ILD progression and survival.
The findings of the present study should be interpreted in the context of specific limitations. There were subtle differences in the baseline characteristics of the SLS I and SLS II cohorts. For instance, the DLCO was lower in SLS I, and this may have been due to less scrupulousness in excluding pulmonary arterial hypertension (PAH). The Baseline Dyspnoea Index (BDI) score was also lower in SLS I, although these differences could be related to using different instruments to administer the BDI in the two studies.29 The quantitative extent of interstitial lung disease/quantitative extent of lung fibrosis in the zone of maximal involvement on HRCT was higher in SLS II, while the quantitative extent of interstitial lung disease in whole lung on HRCT was higher in SLS I. However, in SLS I, non-volumetric CT scans of 1–2 mm slice thickness were acquired at 10 mm increments, while in SLS II volumetric CT scans of 1–1.5 mm slice thickness were acquired contiguously. Overall, the two cohorts were strikingly similar.
A number of SLS I and, to a lesser degree, SLS II participants were lost to follow-up during the course of this long-term follow-up study. This can introduce bias, especially in cases where early censoring occurred. The Cox proportional hazards model was used to deal with time to event data in the presence of censoring. Moreover, while a morbidity and mortality committee adjudicated the causes of death during the 24-month study periods, less detailed information was obtained during long-term follow-up periods regarding causes of death. In addition, although we successfully collected information on the use of immunosuppression following the conclusion of SLS II, the timing, duration and dosages of immunosuppression reported by patients varied widely and precluded any kind of meaningful statistical analysis of these data (in SLS I, very few patients continued on immunosuppression during the second year; beyond this point no data were available regarding immunosuppression use). Finally, while PH was identified as the cause of death in only one of the SLS II patients, this comorbidity may have influenced survival rates in both cohorts.
Notable strengths of the present manuscript include the use of two relatively large, well-characterised SSc-ILD cohorts undergoing standard treatment approaches with uniform follow-up measurements over the course of 2 years. These cohorts comprised patients from multiple SSc Centers of Excellence across the USA, augmenting the generalisability of our study findings to a US population. Furthermore, we identified the same mortality predictor variables in both cohorts, suggesting that our results are likely reproducible in other similar SSc cohorts. Finally, we used a joint model as a means of internal validation.
In summary, the findings of the present analyses demonstrate that increased baseline skin score, increased baseline age, and the course of the FVC and DLCO over 2 years are important predictors of long-term survival in SSc-ILD. Treatment with immunosuppression may not improve long-term survival in patients with SSc-ILD, in contrast to haematopoetic stem cell transplantation,26–28 which seems to offer a more sustained improvement in long-term survival and may especially help those patients who have early, rapidly progressive SSc with organ involvement. Future studies are needed to determine how the duration of immunosuppression affects long-term survival among patients with SSc-ILD. With the emergence of promising new therapies for SSc-ILD (eg, antifibrotics, or combination therapy with antifibrotics and immunosuppression), additional studies are needed to compare how these novel approaches affect survival compared with the current standard of care for SSc-ILD.
Acknowledgments
Bristol-Myers Squibb supplied cyclophosphamide for use in SLS I and Hoffman-La Roche supplied mycophenolate mofetil for use in SLS II. We thank John Dermond and Grace Ibrahim for their assistance in contacting the participants in SLS I and II, respectively.
References
Footnotes
Handling editor Josef S Smolen
Presented at This manuscript was based on work previously published at the following conferences: Systemic Sclerosis World Congress 2018 (Volkmann ER, Tashkin DP, Sim M, et al, The course of the forced vital capacity during treatment for systemic sclerosis-related interstitial lung disease predicts long-term survival in 2 independent cohorts, Journal of Scleroderma and Related Disorders 2018;3(15):69–101) and the American College of Rheumatology Annual Meeting 2017 (Volkmann ER, Tashkin DP, Sim M, et al, The course of the forced vital capacity during treatment for systemic sclerosis-related interstitial lung disease predicts long-term survival in 2 independent cohorts, Arthritis Rheumatol 2017;69(Suppl 10)).
Collaborators The following persons and institutions participated in the Scleroderma Lung Study I: University of California at Los Angeles (UCLA), Los Angeles: PJ Clements, DP Tashkin, R Elashoff, J Goldin, M Roth, D Furst, K Bulpitt, D Khanna, W-LJ Chung, S Viasco, M Sterz, L Woolcock, X Yan, J Ho, S Vasunilashorn, I da Costa; University of Medicine and Dentistry of New Jersey, New Brunswick: JR Seibold, DJ Riley, JK Amorosa, VM Hsu, DA McCloskey, JE Wilson; University of Illinois at Chicago, Chicago: J Varga, D Schraufnagel, A Wilbur, M Lopata, S Arami, P Cole-Saffold; Boston University, Boston: R Simms, A Theodore, P Clarke, J Korn, K Tobin, M Nuite; Medical University of South Carolina, Charleston: R Silver, M Bolster, C Strange, S Schabel, E Smith, J Arnold, K Caldwell, M Bonner; Johns Hopkins School of Medicine, Baltimore: R Wise, F Wigley, B White, L Hummers, M Bohlman, A Polito, G Leatherman, E Forbes, M Daniel; Georgetown University, Washington, DC: V Steen, C Read, C Cooper, S Wheaton, A Carey, A Ortiz; University of Texas at Houston, Houston: M Mayes, E Parsley, S Oldham, T Filemon, S Jordan, M Perry; University of California at San Francisco, San Francisco: K Connolly, J Golden, P Wolters, R Webb, J Davis, C Antolos, C Maynetto; University of Alabama at Birmingham, Birmingham: B Fessler, M Olman, C Sanders, L Heck, T Parkhill; University of Connecticut Health Center, Farmington: N Rothfield, M Metersky, R Cobb, M Aberles, F Ingenito, E Breen; Wayne State University, Detroit: M Mayes, K Mubarak, JL Granda, J Silva, Z Injic, R Alexander; Virginia Mason Research Center, Seattle: D Furst, S Springmeyer, S Kirkland, J Molitor, R Hinke, A Mondt; Data Safety and Monitoring Board: Harvard Medical School, Boston—T Thompson; Veterans Affairs Medical Center, Brown University, Providence, Rhode Island—S Rounds; Cedars Sinai–UCLA, Los Angeles—M Weinstein; Clinical Trials Surveys, Baltimore—B Thompson; Mortality and Morbidity Review Committee: UCLA, Los Angeles—H Paulus, S Levy; Johns Hopkins University, Baltimore—D Martin. The following persons and institutions participated in the Scleroderma Lung Study II: University of Boston, Boston: AC Theodore, RW Simms, E Kissin, FY Cheong; Georgetown University, Washington, DC: VD Steen, CA Read Jr, C Fridley, M Zulmatashvili; Johns Hopkins University, Baltimore: RA Wise, FM Wigley, L Hummers, G Leatherman; Medical University of South Carolina, Charleston: RM Silver, C Strange, FN Hant, J Ham, K Gibson, D Rosson; University of California, Los Angeles (UCLA), Los Angeles: DP Tashkin, RM Elashoff, MD Roth, PJ Clements, D Furst, S Kafaja, E Kleerup, D Elashoff, J Goldin, E Ariola, G Marlis, J Mason-Berry, P Saffold, M Rodriguez, L Guzman, J Brook; University of California, San Francisco (UCSF), San Francisco: J Golden, MK Connolly, A Eller, D Leong, M Lalosh, J Obata; University of Illinois, Chicago: S Volkov, D Schraufnagel, S Arami, D Franklin; Northwestern University, Chicago: J Varga, J Dematte, M Hinchcliff, C DeLuca, H Donnelly, C Marlin; University of Medicine and Dentistry of New Jersey, New Brunswick: DJ Riley, VM Hsu, DA McCloskey; University of Michigan, Ann Arbor: K Phillips, D Khanna, FJ Martinez, E Schiopu, J Konkle; University of Texas, Houston: M Mayes, B Patel, S Assassi, F Tan; National Jewish Health, Denver: A Fischer, J Swigris, R Meehan, K Brown, T Warren, M Morrison; University of Utah, Salt Lake City: MB Scholand, T Frecht, P Carey, M Villegas; University of Minnesota, Minneapolis: J Molitor, P Carlson.
Contributors All coauthors met the criteria for authorship.
Funding This work was supported in part by the NIH/NIAID: N01-AI05419 (KMS) and HHSN-272201100025C (KMS); the Scleroderma Foundation (ERV); NHI/NIAMS: R01 AR 070470 and K24 AR 063120 (DK); and NHLBI/NIH: R01 HL089758 (DPT), R01 HL089901 (RME), U01 HL 60587 (DPT) and U01 HL 60606 (RME).
Competing interests None declared.
Patient consent Not required.
Ethics approval UCLA IRB and the institutional review board of each site approved the studies.
Provenance and peer review Not commissioned; externally peer reviewed.