Article Text

Abstract
Objectives To evaluate the effect of subcutaneous (s.c.) secukinumab, an interleukin-17A inhibitor, on clinical signs and symptoms and radiographic progression in patients with psoriatic arthritis (PsA).
Methods Adults (n=996) with active PsA were randomised 2:2:2:3 to s.c. secukinumab 300 mg or 150 mg with loading dose (LD), 150 mg without LD or placebo. All groups received secukinumab or placebo at baseline, weeks 1, 2 and 3 and then every 4 weeks from week 4. The primary endpoint was the proportion of patients achieving an American College of Rheumatology 20 (ACR20) response at week 16.
Results Significantly more patients achieved an ACR20 response at week 16 with secukinumab 300 mg with LD (62.6%), 150 mg with LD (55.5%) or 150 mg without LD (59.5%) than placebo (27.4%) (p<0.0001 for all; non-responder imputation). Radiographic progression, as measured by van der Heijde-modified total Sharp score, was significantly inhibited at week 24 in all secukinumab arms versus placebo (p<0.01 for 300 mg with LD and 150 mg without LD and p<0.05 for 150 mg with LD; linear extrapolation). Adverse event rates at week 24 were similar across treatment arms: 63.1% (300 mg with LD), 62.7% (150 mg with LD), 61.1% (150 mg without LD) and 62.0% (placebo). No deaths or new safety signals were reported.
Conclusion S.c. secukinumab 300 mg and 150 mg with and without LD significantly improved clinical signs and symptoms and inhibited radiographic structural progression versus placebo at week 24 in patients with PsA.
Trial registration number NCT02404350; Results.
- dmards (biologic)
- cytokines
- treatment
- psoriatic arthritis
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Psoriatic arthritis (PsA) is a chronic, inflammatory disease characterised by peripheral arthritis, axial disease, dactylitis, enthesitis and skin and nail psoriasis.1 2 It can have a substantial impact on quality of life and work productivity,3 with many patients experiencing irreversible joint damage and disability.1 4 Indeed, bone erosions occur in approximately half of patients within 2 years.5
Enhanced understanding of the pathophysiology of PsA has aided the development of targeted therapies to manage its signs and symptoms. The proinflammatory cytokine interleukin (IL)-17A mediates multiple biological functions that result in joint and entheseal inflammation, damage and tissue remodelling, which are characteristic of PsA.6–8 Recommendations from the European League Against Rheumatism (EULAR)9 and the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA)10 recognise targeting IL-17A as a therapeutic strategy to manage all the main clinical domains of PsA.
Secukinumab, a fully human monoclonal antibody that selectively neutralises IL-17A,11 is approved in many countries for the treatment of PsA, psoriasis and ankylosing spondylitis. There is now an extensive body of evidence involving more than 2700 patients demonstrating the efficacy of secukinumab in PsA.12–18 Data from the phase III studies, FUTURE 1 and FUTURE 2, have shown that secukinumab provides rapid and significant improvements in the signs and symptoms of PsA that are sustained for up to 3 years of therapy.12–18 These clinical benefits have been observed in patients naïve to biological therapy and in those with an intolerance or inadequate response to agents targeting tumour necrosis factor (TNF).9 10 13–18 Data from FUTURE 1 have shown that secukinumab significantly inhibits joint structural damage through 24 weeks,13 with benefits maintained out to 2 years.17 FUTURE 1 used an intravenous loading followed by subcutaneous (s.c.) dosing regimen for secukinumab and did not evaluate a dose higher than 150 mg.13 FUTURE 2 used s.c. loading and maintenance dosing of secukinumab 300, 150 and 75 mg14 (the 300 and 150 mg dosing regimens were subsequently adopted as the approved regimens in PsA), but it did not examine radiographic progression and, until now, these data have been lacking. Here, we report primary results from the ongoing FUTURE 5 study, the largest randomised, controlled study to date of a biologic in PsA. The study was designed to evaluate the impact of s.c. secukinumab 300 and 150 mg on clinical signs and symptoms and radiographic progression as well as evaluating the short-term benefit of the loading regimen. This trial is ongoing and will provide long-term data out to 2 years.
Methods
Study design and participants
FUTURE 5 (NCT02404350) is a randomised, double-blind, placebo-controlled, parallel-group phase III trial. The study design is shown in online supplementary figure 1. The study was conducted in accordance with the principles of the Declaration of Helsinki.19 Patients provided written informed consent before study-related procedures were undertaken.
Supplemental material
Patients were aged ≥18 years and met the ClASsification criteria for Psoriatic ARthritis2 at screening, with symptoms of moderate-to-severe PsA for at least 6 months: ≥3 tender joints and ≥3 swollen joints despite ≥4 weeks of treatment with non-steroidal anti-inflammatory drugs (NSAIDs), or an intolerance to them, and active or documented history of plaque psoriasis or psoriatic nail changes. Concomitant corticosteroids (≤10 mg/day prednisone or equivalent), NSAIDs and methotrexate (≤25 mg/week) were allowed, provided the dose was stable and remained so for the first 24 weeks of the study. Patients who had previously used anti-TNF agents could enrol if they had experienced an inadequate response or stopped treatment due to safety or intolerance (anti-TNF-IR). Patients taking anti-TNF therapy and/or a disease-modifying antirheumatic drug other than methotrexate required a washout ranging from 4 to 10 weeks before randomisation, depending on the prohibited treatment used. Key exclusion criteria included active/history of ongoing infection, prior use of a biologic other than an anti-TNF agent, use of ≥3 anti-TNF agents and active inflammatory disease other than PsA.
Treatment and randomisation
Following a screening period of up to 10 weeks, Interactive Response Technology was used to randomly assign eligible patients in a 2:2:2:3 ratio to one of four treatment groups: secukinumab 300 mg with loading dose (LD), secukinumab 150 mg with LD, secukinumab 150 mg without LD or placebo, all administered s.c. (online supplementary figure 1). Patients self-administered their own treatment using prefilled syringes at baseline, weeks 1, 2 and 3 followed by treatment every 4 weeks from week 4. Patients in the secukinumab 150 mg without LD arm were administered placebo at weeks 1, 2 and 3 to conceal treatment allocation. At week 16, patients in the placebo arm with <20% improvement from baseline in tender and swollen joint counts (SJCs) were switched in a double-blind manner to receive s.c. secukinumab 300 mg or 150 mg, preassigned at original randomisation. Remaining patients in the placebo arm were switched to blinded secukinumab 300 mg or 150 mg at week 24.
Randomisation was stratified according to previous anti-TNF therapy use, with patients being anti-TNF-naïve (planned enrolment about 70%) or anti-TNF-IR. Patients, investigators and assessors remain masked to the treatment assignment until all patients reach week 52.
Outcomes
The primary efficacy endpoint was the proportion of patients with an American College of Rheumatology 20 (ACR20) response at week 16. The key secondary hierarchical endpoint was radiographic structural progression at week 24, as measured by change from baseline in van der Heijde-modified total Sharp score (vdH-mTSS), which ranges from 0 to 528 (higher scores indicating more articular damage)20 based on independent assessments of hand/wrist/foot radiographs obtained at baseline, week 16 (non-responders) and week 24 by two central blinded readers (plus an adjudicator if required). Other hierarchical secondary endpoints were assessed at week 16 and included: proportion of patients achieving a 75% or 90% improvement from baseline in the Psoriatic Area and Severity Index (PASI75 and PASI90, respectively);21 proportion of patients with an ACR50 response; change from baseline on the Health Assessment Questionnaire-Disability Index (HAQ-DI scores range from 0 to 3 with higher scores indicate greater disability);22 change from baseline in the 28-joint Disease Activity Score using C reactive protein (DAS28-CRP, with higher scores indicating more active disease);23 and resolution of enthesitis and dactylitis. Definitions of the ACR20 response and secondary points used in the study are provided in the online supplementary methods.
Supplemental material
Prespecified exploratory endpoints of ACR70 response, the proportion of patients with no structural progression (change from baseline in vdH-mTSS ≤0.5) at week 24 and both the primary endpoint (ACR20 at week 16), change from baseline vdH-mTSS at week 24 by prior use of anti-TNF therapy and the proportion of patients achieving minimal disease activity (MDA) are also reported. MDA is assessed as five of the seven following: ≤1 tender joint count, ≤1 SJC, PASI≤1 or body surface area (BSA)≤3%, patient pain visual analogue score (VAS)≤15, patient global assessment of disease activity VAS≤20, HAQ-DI≤0.5, tender entheseal points≤1.24 Disease Activity index for PSoriatic Arthritis (DAPSA) was analysed posthoc. DAPSA is a continuous index, calculated by summing of individual scores for: (1) tender joint count (TJC), (2) SJC, (3) patient global assessment, (4) patient assessment of pain and (5) CRP level, using 66/68 joint counts.25 DAPSA ranges from 0 to 164; DAPSA remission (REM; range 0–4) and low disease activity (LDA; range 5–14) were assessed. Overall safety and tolerability of secukinumab over the 24 weeks was assessed by monitoring adverse events (AEs), serious AEs (SAEs), laboratory assessments and vital signs.
Statistical analyses
Sample sizes were calculated based on an overall two-sided 5% type I error rate. As three secukinumab regimens were tested versus placebo for ACR20, the type I error was separated into a 1.67% two-sided for each comparison. Based on ACR20 response rates of previous studies,14 a sample size of 220 patients in each secukinumab group, and 330 in the placebo group was estimated to provide around 99% power (for all three dose regimens) to detect a treatment difference in the primary endpoint (ACR20 at week 16) based on Fisher’s exact test.
For the key secondary endpoint, radiographic structural progression (vdH-mTSS), a treatment difference of 0.52 and SD of 1.13 with active treatment and 2.44 with placebo were observed in historical data.13 Based on these assumptions, there is 83% power to detect a treatment difference using Satterthwaite t-test.
A sequential hierarchical testing method (online supplementary figure 2) was used to maintain the familywise type I error rate at 5% across the primary and ranked secondary specified endpoints. P values were calculated as 2-sided. Patients were analysed according to randomised treatment.
Supplemental material
Statistical analyses were based on logistic regression for binary efficacy variables (eg, ACR20/50/70 and so on), non-parametric analysis of covariance for radiographic data (if baseline and ≥1 postbaseline radiographic assessments were available) and mixed-effects models for repeated measures (MMRM) for continuous variables (eg, DAS28-CRP, HAQ-DI). All models fitted included anti-TNF status, weight and the corresponding baseline value as a covariate as well as treatment as a factor (time, treatment by time and baseline by time interaction were also used for MMRM models). Missing values and placebo patients rescued at week 16 were imputed as non-responders for binary endpoints (rescue penalty), linear extrapolation was applied for radiographic data (if baseline and week 16 values were available) and the missing at random assumption of the MMRM analysis was applied for continuous endpoints.
Safety endpoints were assessed for all patients who received ≥1 dose of study drug and were summarised descriptively. Patients were evaluated according to the treatment they received.
Results
Patients
A total of 996 patients were randomly assigned to receive secukinumab 300 mg with LD (n=222), secukinumab 150 mg with LD (n=220), secukinumab 150 mg without LD (n=222) or placebo (n=332). At week 24, a total of 66 (6.9%) patients had discontinued, with the greatest number (n=37) coming from the placebo group. The most common reason for discontinuation was patient/guardian decision (3.2% overall; 5.7% placebo, 3.2% 150 mg without LD and 1.4% for both 150 mg and 300 mg with LD) followed by AEs (1.6% overall; 2.7% placebo, 0.9% 150 mg each with and without LD, and 1.4% 300 mg with LD). Patient disposition is shown in online supplementary figure 3.
Supplemental material
Demographics and baseline characteristics were balanced between treatment arms (table 1). Overall, the mean age was 48.8 years, 49.8% were female and the mean time since PsA diagnosis was 6.6 years. At baseline, 70.4% of patients were anti-TNF-naïve and 50.1% were receiving concomitant methotrexate. Approximately half (51.6%) of the patients had psoriasis affecting ≥3% of their BSA. Enthesitis was present in 60.4% and dactylitis in 39.1% of patients. Mean swollen and tender joint counts were 11.5 and 21.0, respectively and mean HAQ-DI score was 1.3.
Demographics and baseline characteristics for the randomised set
Efficacy
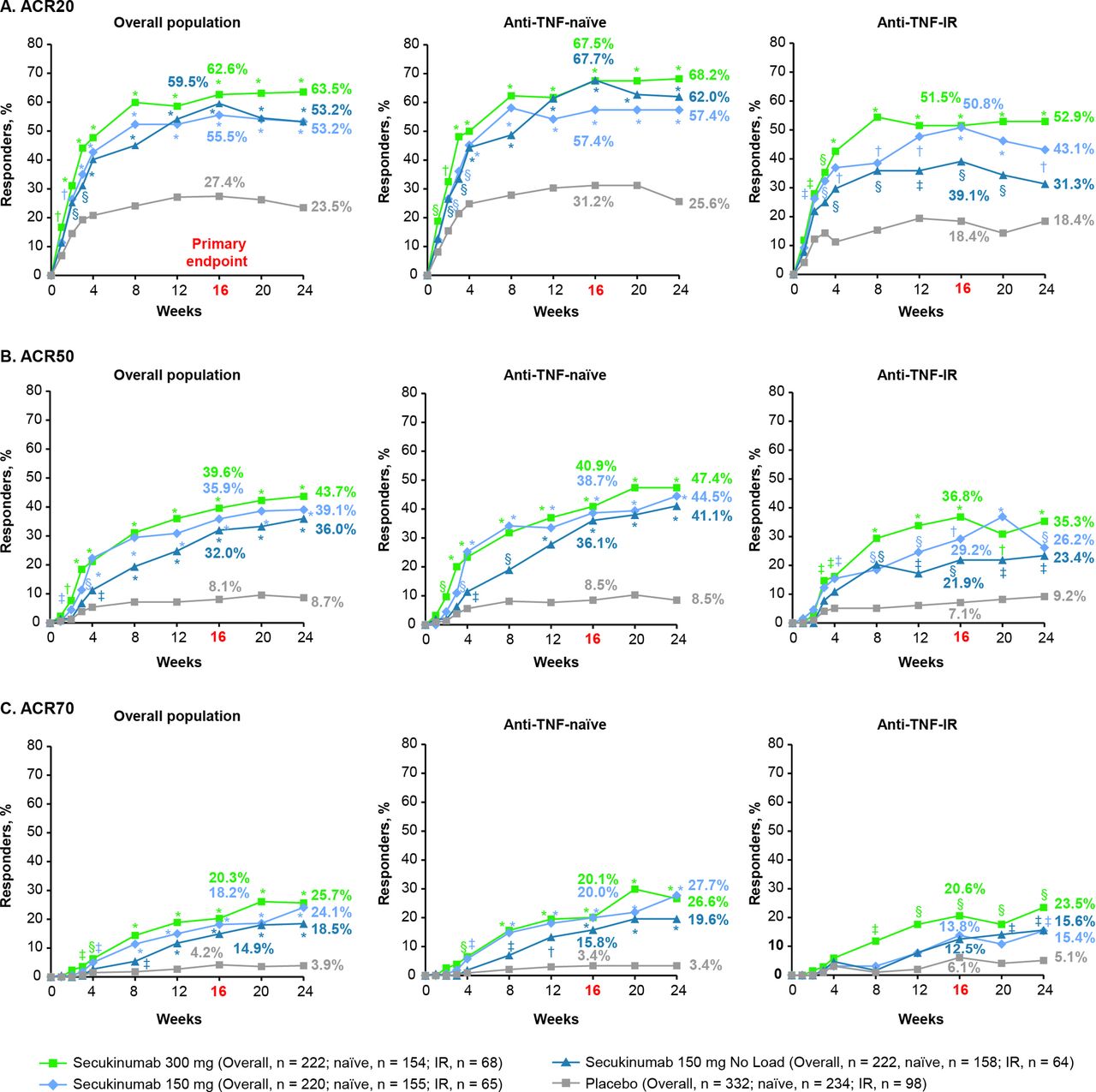
The primary endpoint was met with all secukinumab doses. ACR20 response rates at week 16 were significantly higher with secukinumab 300 mg with LD (62.6%), 150 mg with LD (55.5%) or 150 mg without LD (59.5%) than placebo (27.4%; p<0.0001 for all doses vs placebo; figure 1, online supplementary table 1 and table 2). ACR50/70 response rates at week 16 were also significantly higher with all secukinumab doses versus placebo (figure 1 and online supplementary table 1). ACR20/50/70 response rates at week 16 were numerically higher in anti-TNF-naive than anti-TNF-IR patients for all secukinumab doses. All secondary hierarchical endpoints were significant for secukinumab versus placebo at week 16, except for enthesitis and dactylitis resolution for the 150 mg without LD group (table 2, figure 2 and online supplementary table 2).
Supplemental material
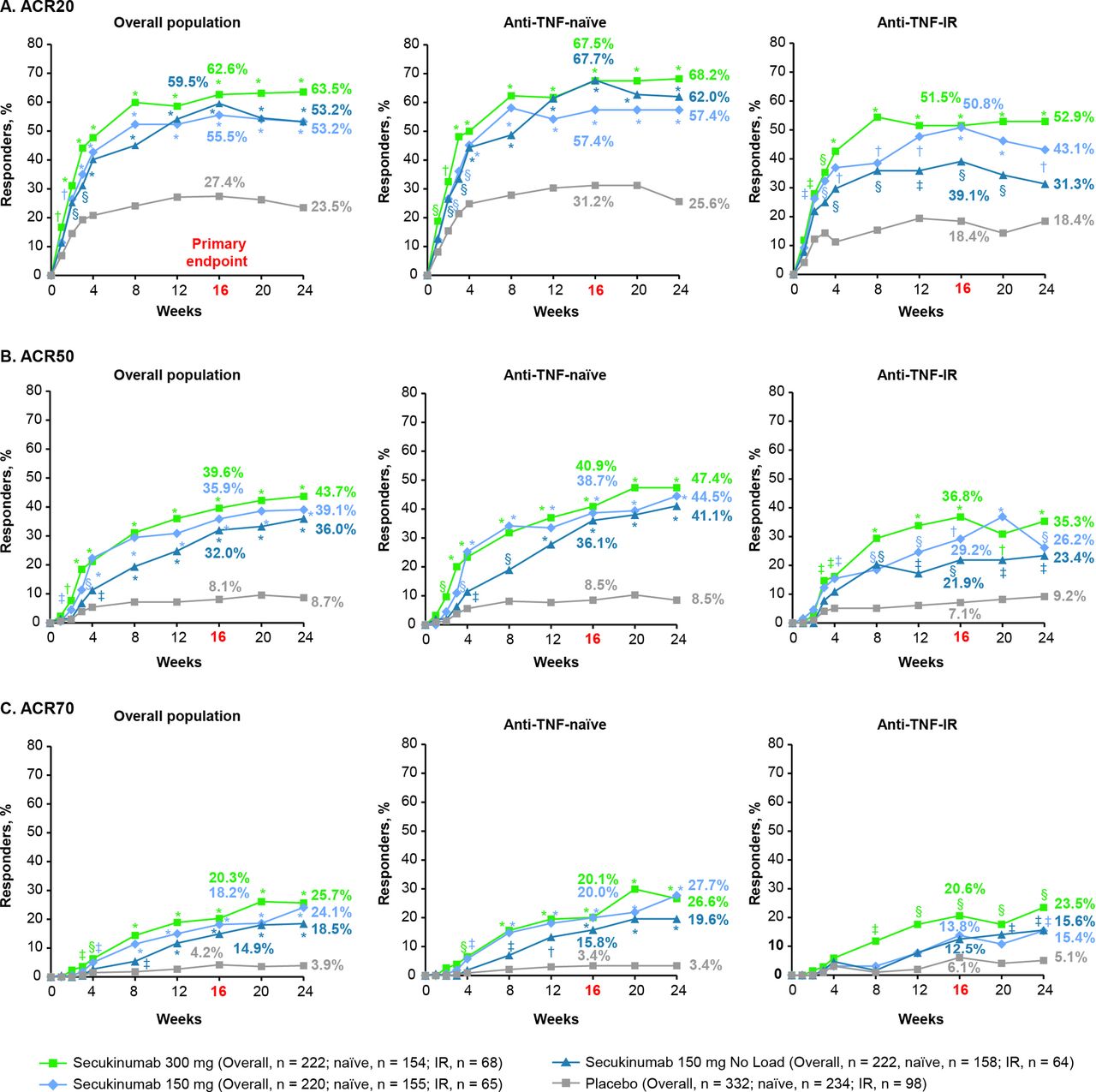
(A) ACR20, (B) ACR50 and (C) ACR70 response rates from baseline up to week 24a in the overall population and by anti-TNF status. *P<0.0001; †p<0.001; §p<0.01; ‡p<0.05 unadjusted p values versus placebo (Statistical analysis was based on logistic regression. Missing values and placebo patients rescued at week 16 were imputed as non-responders.) aThe primary endpoint was ACR20 response in the overall population at week 16. ACR20/50/70, ≥20/50/70% improvement from baseline in American College of Rheumatology response criteria; anti-TNF-IR, intolerance or inadequate response to antitumour necrosis factor therapy.

Resolution of enthesitis and dactylitis in the overall population from baseline up to week 24a.*P<0.0001; †p<0.001; §p<0.01; ‡p<0.05 unadjusted p values versus placebo. (Statistical analysis was based on logistic regression. Missing values and placebo patients rescued at week 16 were imputed as non-responders.) aResolution of dactylitis and enthesitis were not significant for secukinumab 150 mg without load in hierarchical testing.
Comparison of secukinumab versus placebo at week 16 for prespecified hierarchical endpoints
Mean changes from baseline in vdH-mTSS demonstrated significant inhibition of radiographic structural progression at week 24 in all secukinumab groups versus placebo: 0.08 (300 mg with LD; p<0.01), 0.17 (150 mg with LD; P<0.05), –0.09 (150 mg without LD; p<0.05) versus 0.50 (placebo; figure 3, online supplementary table 3 and table 2). The proportion of patients with no radiographic structural progression at week 24, defined as ≤0.5 change from baseline in vdH-mTSS, was higher across all secukinumab dose regimens than placebo: 191/217 (88.0%) patients in the secukinumab 300 mg with LD group, 170/213 (79.8%) in the 150 mg with LD group and 176/210 (83.8%) in the 150 mg without LD group versus 218/296 (73.6%) in the placebo group (online supplementary figure 4).

![[SP2.jpg]](https://ard.bmj.com/content/annrheumdis/77/6/890/DC1/embed/inline-supplementary-material-1.jpg?download=true){kind=link}
![[SP3.jpg]](https://ard.bmj.com/content/annrheumdis/77/6/890/DC3/embed/inline-supplementary-material-3.jpg?download=true){kind=link}
![[SP4.jpg]](https://ard.bmj.com/content/annrheumdis/77/6/890/DC4/embed/inline-supplementary-material-4.jpg?download=true){kind=link}
{kind=link}
{kind=link}
{kind=link}
Change in vdH-mTSS from baseline at week 24 (non-parametric ANCOVA-linear extrapolation in the overall population and by anti-TNF status. †P<0.001; §p<0.01; ‡p<0.05 unadjusted p values versus placebo (Statistical analysis was based on a non-parametric ANCOVA. Linear extrapolation was applied if a baseline and week 16 value were available). ANCOVA, analysis of covariance; anti-TNF-IR, intolerance or inadequate response to antitumour necrosis factor therapy; LD, loading dose; vdH-mTSS, van der Heijde-modified total Sharp score.
The 300 mg secukinumab dose provided numerically better efficacy in all hierarchical endpoints versus the 150 mg regimens, with the strongest treatment difference observed for psoriasis improvement. PASI 75 response rates at week 16 were 70.0% with 300 mg with LD, 60.0% with 150 mg with LD, 58.1% with 150 mg without LD and 12.3% with placebo; corresponding PASI 90 responses rates were 53.6%, 36.8%, 31.6% and 9.3%, respectively. Treatment responses were greater in anti-TNF-naïve patients than in anti-TNF-IR patients (Figures 1,3). In the 150 mg groups, patients receiving LD generally had an earlier onset of response and numerically greater efficacy versus patients without LD (figure 1).
The proportion of patients achieving MDA at week 16 were 33.0% with secukinumab 300 mg, 28.2% with 150 mg with LD, 23.0% with 150 mg without LD and 8.4% with placebo (p<0.001 for all secukinumab doses versus placebo; online supplementary figure 5). The proportion of patients achieving DAPSA REM/LDA states at week 16 were 15.2%/37.4% with secukinumab 300 mg, 13.4%/27.8% with 150 mg with LD, 9.0%/34.8% with 150 mg without LD and 2.3%/18.9% with placebo (online supplementary figure 5).
Supplemental material
![[SP8.jpg]](https://ard.bmj.com/content/annrheumdis/77/6/890/DC6/embed/inline-supplementary-material-6.jpg?download=true){kind=link}
Safety
Exposure and safety are reported in table 3. The duration of exposure over the 24 weeks was higher for secukinumab (329.7 patient-years) than placebo (122.7 patient-years) due to 158 patients in the placebo group switching to secukinumab at week 16. The most commonly reported AEs represented upper respiratory tract infections; other common AEs are reported in table 3. No deaths or major adverse cardiac events were reported. Non-fatal SAE rates were low overall and similar for secukinumab (3.0%) and placebo (3.6%). Most SAEs represented single events with no discernible pattern.
Exposure and rates of deaths, discontinuations, AEs, SAEs and selected AEs and SAEs of interest up to week 24†
Selected SAEs of interest included: one report of suicidal thoughts in a patient with a history of anxiety who continued in the study without further episodes; one anaphylactic reaction after the second secukinumab dose, which resulted in patient discontinuation; one new diagnosis of ulcerative colitis in a secukinumab patient with no prior gastrointestinal medical history and who remained in the study and one case of Crohn’s disease in a patient with a history of colitis. There was one mild, non-serious exacerbation of Crohn’s disease in a secukinumab-treated patient who continued on study treatment and remained in the study; the event was resolved at the time of reporting.
Reports of Candida infections included: one case of oral thrush (300 mg with LD), four cases of oral candidiasis (one in 150 mg with LD; two in 300 mg with LD; one in placebo) and four cases of vulvovaginal candidiasis (two in 150 mg with LD; one in 300 mg; one in placebo). These were of mild severity, except one moderately severe vulvovaginal candida infection; all resolved with standard therapy. No systemic fungal infections or newly diagnosed tuberculosis infections were reported, and incidences of injection site reactions were low across all groups.
Discussion
FUTURE 5 is the largest randomised phase III trial to date of a biologic in PsA. In this study, s.c. administration of secukinumab 300 mg and 150 mg provided rapid and significant improvement versus placebo in most clinical domains of PsA and inhibited radiographic progression at week 24.
The primary endpoint, ACR20 response at week 16, was met for all secukinumab regimens, and secondary endpoints were significant for all secukinumab doses except for enthesitis and dactylitis resolution in the 150 mg without LD group. These results confirm and extend previous findings relating to the efficacy of secukinumab in PsA.12–18 In addition, clinical response rates (ACR20/50/70) at week 16 were higher in anti-TNF-naive patients than in those who were anti-TNF-IR for all secukinumab doses.
These data provide the first evidence that s.c. secukinumab loading and maintenance dosing regimens and the higher dose of 300 mg significantly inhibit joint structural damage in PsA. More patients who received secukinumab versus those who received placebo had no radiographic progression through week 24. Inhibition of radiographic progression was observed in both anti-TNF-naïve and anti-TNF-IR patients, although statistical significance was not reached in the anti-TNF-IR population. The lack of significance could be affected by a number of factors, including the relatively small number of anti-TNF-IR patients and the heterogeneity of this subpopulation, which comprised patients who previously failed anti-TNF treatment for any one of several reasons, including lack of primary or secondary efficacy, intolerance or safety concerns.26 This study was limited in that it was not designed to identify a difference between doses or to assess differences in response according to previous anti-TNF use.
Patients enrolled in this study had a long duration of disease (mean around 6.5 years) and very active disease as evidenced by the relatively high tender and swollen joint scores and the large proportion of patients with enthesitis and dactylitis at baseline. Additionally, around 30% of patients had previously received one or more anti-TNF treatments before entering this trial. While anti-TNF agents have been shown to improve outcomes in PsA,27–31 many patients experience inadequate disease control, treatment intolerance or loss of response over time.1 32 In agreement with previous studies,9 10 13–18 secukinumab treatment in FUTURE 5 was shown to be efficacious in both anti-TNF-naïve and anti-TNF-IR patients, with clinical responses generally being higher in anti-TNF-naïve patients. These results confirm that secukinumab is a suitable treatment option for biologic-naïve patients and those who have previously failed anti-TNF therapy.
The secukinumab 300 mg dose consistently provided numerically better responses versus the 150 mg dose, with or without LD, in clinical endpoints such as ACR20/50/70, resolution of enthesitis and dactylitis, HAQ-DI, DAS28-CRP, PASI 75 and PASI 90, particularly in anti-TNF-IR patients. Data from FUTURE 5 also demonstrate the benefit of a loading regimen in terms of providing an earlier onset of action, especially on higher efficacy endpoints such as ACR50/70, dactylitis and in the treatment of psoriasis symptoms, as indicated by PASI 75 and PASI 90, and in the proportion of patients achieving MDA and DAPSA-REM.
The safety profile was consistent with that previously reported for PsA12–18 and psoriasis.32 The types and incidences of most AEs with secukinumab were similar to those for placebo for the entire 24-week treatment period, without evidence of dose-dependency. The rate of Candida infections was higher with secukinumab treatment than with placebo; this is consistent with previous reports13 14 and considered to be related to the role of IL-17 in mucocutaneous defence against Candida infections.33 No systemic fungal infections were reported. All cases of Candida infection resolved with standard oral therapy and patients continued in the study.
This study demonstrated that s.c. secukinumab 300 mg and 150 mg provided significant inhibition of radiographic progression, and has demonstrated the benefit of a LD regimen (regardless of the dose), particularly when aiming for higher levels of response and faster outcomes in joint and skin endpoints. The safety profile of secukinumab was consistent with previous reports, with no new safety signals observed.12–18 FUTURE 5 confirms and extends the results of previous data demonstrating the efficacy of s.c. administration of secukinumab in achieving comprehensive treatment goals in PsA.
Supplemental material
![[SP5.jpg]](https://ard.bmj.com/content/annrheumdis/77/6/890/DC7/embed/inline-supplementary-material-7.jpg?download=true){kind=link}
Supplemental material
![[SP6.jpg]](https://ard.bmj.com/content/annrheumdis/77/6/890/DC8/embed/inline-supplementary-material-8.jpg?download=true){kind=link}
Supplemental material
![[SP7.jpg]](https://ard.bmj.com/content/annrheumdis/77/6/890/DC9/embed/inline-supplementary-material-9.jpg?download=true){kind=link}
References
Footnotes
Handling editor Josef S Smolen
Contributors All authors were involved in the study design and/or collection, analysis and interpretation of the data, provided critical revision of the manuscript and approved the final version to be submitted for publication.
Funding This analysis was supported by Novartis Pharmaceuticals Corporation. Medical writing support was provided by Gillian Brodie, MSc and Martin Wallace, PhD of Novartis Ireland Ltd., Dublin, Ireland, which was funded by Novartis Pharmaceuticals Corporation in accordance with Good Publication Practice (GPP3) guidelines.
Competing interests PM: Research grants from AbbVie, Amgen, BMS, Celgene, Janssen, Lilly, Novartis, Pfizer, SUN and UCB; consulting fees from AbbVie, Amgen, BMS, Celgene, Covagen, Crescendo, Janssen, LEO, Lilly, Merck, Novartis, Pfizer, SUN and UCB; speakers’ bureau for AbbVie, Amgen, BMS, Celgene, Genentech, Janssen, Lilly, Pfizer and UCB. DvdH: Consulting fees AbbVie, Amgen, Astellas, AstraZeneca, BMS, Boehringer Ingelheim, Celgene, Daiichi, Eli-Lilly, Galapagos, Gilead, Glaxo-Smith-Kline, Janssen, Merck, Novartis, Pfizer, Regeneron, Roche, Sanofi, Takeda, UCB. Director of Imaging Rheumatology. RL: Consultation or participation in advisory boards: Abbott/AbbVie, Ablynx, Amgen, Astra-Zeneca, Bristol-Myers Squibb, Centocor, GlaxoSmithKline, Novartis, Merck, Pfizer, Roche, Schering- Plough, UCB, Wyeth. Research grants: Abbott, Amgen, Centocor, Novartis, Pfizer, Roche, Schering-Plough, UCB, Wyeth. Speaker fees: Abbott, Amgen, Bristol-Myers Squibb, Centocor, Merck, Pfizer, Roche, Schering-Plough, UCB, Wyeth. RL: Director of Rheumatology Consultancy BV, which is a registered company under Dutch law. SM: Employee of Novartis, with Novartis stock. PR: Consulting fees for Abbott, AbbVie, Amgen, BMS, Celgene, Janssen, Novartis, Pfizer and Roche. Consultant to pharmaceutical companies dealing with biologic agents in rheumatology. HT: Consultation or participation in advisory boards: Abbvie, Novartis, Pfizer, UCB, Eli-Lilly, Janssen Education Grants: Novartis, Pfizer. AS: Research/Clinical trial grants from AbbVie, Gilead, Sanofi, Regeneron, Amgen, Roche, BMS, Janssen, Lilly, Novartis, Pfizer, UCB, Astra Zeneca, MedImmune, FujiFilm, Nichi-Iko, Mallinckrodt. Speakers’ bureau for AbbVie. EB: Consulting and speaking fees: Amgen, Roche, Eli Lilly, Pfizer, MSD, Novartis. SN: Consulting and speaker fees: Pfizer, Novartis, Astra-Zeneca, Janssen, Astellas, Roche. KM: Employee of Novartis, without Novartis stock. AR: Employee of Novartis, with Novartis stock. LP: Employee of Novartis, with Novartis stock. KA: Employee of Novartis, with Novartis stock.
Patient consent Patients provided written informed consent before study-related procedures were undertaken.
Ethics approval Independent ethics committees or institutional review boards of participating centers.
Provenance and peer review Not commissioned; externally peer reviewed.
Correction notice This article has been corrected since it published Online First. Tables 1 and 2 have been amended.