Article Text

Abstract
Objective Interleukin (IL)-17A has emerged as pivotal in driving tissue pathology in immune-mediated inflammatory diseases. The role of IL-17F, sharing 50% sequence homology and overlapping biological function, remains less clear. We hypothesised that IL-17F, together with IL-17A, contributes to chronic tissue inflammation, and that dual neutralisation may lead to more profound suppression of inflammation than inhibition of IL-17A alone.
Methods Preclinical experiments assessed the role of IL-17A and IL-17F in tissue inflammation using disease-relevant human cells. A placebo-controlled proof-of-concept (PoC) clinical trial randomised patients with psoriatic arthritis (PsA) to bimekizumab (n=39) or placebo (n=14). Safety, pharmacokinetics and clinical efficacy of multiple doses (weeks 0, 3, 6 (240 mg/160 mg/160 mg; 80 mg/40 mg/40 mg; 160 mg/80 mg/80 mg and 560 mg/320 mg/320 mg)) of bimekizumab, a humanised monoclonal IgG1 antibody neutralising both IL-17A and IL-17F, were investigated.
Results IL-17F induced qualitatively similar inflammatory responses to IL-17A in skin and joint cells. Neutralisation of IL-17A and IL-17F with bimekizumab more effectively suppressed in vitro cytokine responses and neutrophil chemotaxis than inhibition of IL-17A or IL-17F alone. The PoC trial met both prespecified efficacy success criteria and showed rapid, profound responses in both joint and skin (pooled top three doses vs placebo at week 8: American College of Rheumatology 20% response criteria 80.0% vs 16.7% (posterior probability >99%); Psoriasis Area and Severity Index 100% response criteria 86.7% vs 0%), sustained to week 20, without unexpected safety signals.
Conclusions These data support IL-17F as a key driver of human chronic tissue inflammation and the rationale for dual neutralisation of IL-17A and IL-17F in PsA and related conditions.
Trial registration number NCT02141763; Results.
- psoriatic arthritis
- inflammation
- cytokines
- autoimmune diseases
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Pro-inflammatory cytokines such as tumour necrosis factor (TNF) and interleukin 6 (IL-6) are key drivers of chronic tissue inflammation and are well-validated therapeutic targets in a variety of immune-mediated inflammatory diseases.1–3 More recently, IL-17A was identified as a driver of joint and skin inflammation, with preclinical promise translating to success with anti-IL-17A inhibitors for the treatment of diseases such as psoriasis (PSO), psoriatic arthritis (PsA) and ankylosing spondylitis.4 5 Nevertheless, complete remission remains rare and many patients respond only partially, or not at all, to treatment blocking these cytokines. Therefore, we hypothesised that novel dual-cytokine blockade may have a more profound impact on chronic tissue inflammation than targeting IL-17A alone.
IL-17A shares greater than 50% structural homology and overlapping biological function with another IL-17 family member, IL-17F.6 7 Both cytokines are expressed by the same cell types, including Th17 cells, γδ-T cells and innate lymphoid cells; can be secreted as homodimers or IL-17A–IL-17F heterodimers;8 9 and signal through the same IL-17RA/RC receptor complex. IL-17A and IL-17F are both upregulated in a variety of inflamed human tissues10–13 and co-operate with other pro-inflammatory cytokines, such as TNF, to amplify inflammatory responses.14 Together, these observations led us to hypothesise that IL-17F, in addition to IL-17A, contributes to chronic tissue inflammation in humans, and that dual neutralisation of IL-17A and IL-17F could result in a more profound suppression of inflammation both in vitro and in vivo.
We tested this hypothesis in vitro using human joint and skin cells, and we also conducted a proof-of-concept (PoC) trial in patients with PsA, evaluating bimekizumab (formerly UCB4940), a humanised monoclonal IgG1 antibody (mAb) that potently and selectively neutralises the biological function of both human IL-17A and IL-17F, versus placebo.
Methods
Preclinical methods
mRNA expression analysis
Expression of IL-17A and IL-17F was assessed in the synovial tissue of patients with PsA by duplex qPCR analysis using TaqMan gene expression assays (ThermoFisher) for IL-17A (Hs00174383_m1), IL-17F (Hs00369400_m1) and GAPDH (4310884E) according to the manufacturer’s protocol (online supplementary information).
Supplementary file 1
Characterisation of the anti-IL-17 antibodies’ affinities for human IL-17A and IL-17F
Biomolecular interaction analysis was performed using a Biacore 3000 (Biacore AB). Bimekizumab and the anti-IL-17A antibody were affinity-matched for binding IL-17A whereas bimekizumab has a higher affinity for IL-17F than the anti-IL-17F antibody (generated in house at UCB Pharma); however, it was used at maximal concentrations in all blockade assays (online supplementary table s1).
In vitro effects of recombinant IL-17F on normal human dermal fibroblasts and synoviocytes from patients with psoriatic arthritis
Cells (online supplementary information) were stimulated for 24 hours with recombinant IL-17A (50 ng/mL), IL-17F (50 ng/mL) or both (50 ng/mL each) in the presence of TNF (1 ng/mL). In synoviocytes from patients with PsA, protein levels of secreted IL-6, IL-8 and MMP3 were measured using standard ELISA technique (kits CT205, Ucyteck, the Netherlands (IL-6); CT212, Ucyteck, the Netherlands (IL-8); DY513, R&D systems, UK (MMP3)). In normal human dermal fibroblasts (NHDFs), protein levels of secreted IL-8 were measured by Luminex (R&D Systems, Cat No. LUH208).
Synoviocyte/primary normal human dermal fibroblasts blockade experiments
Blood CD4+CD45RO+CCR6+CXCR3− Th17 cells sorted from healthy donors were stimulated with anti-CD3 and anti-CD28 for 96 hours (online supplementary information). Th17 supernatant was added (as 1:10 dilution) to synoviocytes or NHDFs (online supplementary information) alone, or in combination with 10 µg/mL anti-IL-17A, anti-IL-17F, bimekizumab, anti-TNF or A33 IgG control antibody for 24 hours.
Pathway-focused gene array was conducted in normal synoviocytes and primary NHDFs to profile the expression of 360 inflammatory cytokines, chemokines and their receptors, as well as genes involved in cytokine–cytokine receptor interactions, many of which are linked to IL-17/IL-23 biology, including IL-6 and IL-8. Inflammatory mediator levels were quantified using a Luminex 30-plex cytokine/chemokine array (Thermo Fisher LHC6003) according to the manufacturer’s protocol.
Chemotaxis assay
Neutrophils were derived from whole blood from healthy donors following a red cell lysis step (Thermofisher A1049201) then resuspended at a concentration of 107 cells/mL in DMEM; 200 mL was applied to the upper chamber of a 5-µm transwell (24-well format), 6.5 mm membrane. Diluted Th17 supernatant (500 µL) was placed in the lower transwell chamber to act as chemo-attractant. Co-cultures were incubated for 5 hours at 37°C in a 5% CO2 culture incubator and migrated neutrophils (CD18 FITC) were enumerated using flow cytometry (BD Fortessa X20). Further details on the chemotaxis assay are provided in the online supplementary information.
Clinical methods
Study design
PA0007 was a phase 1b, randomised, double-blind, placebo-controlled, PoC study evaluating the safety, pharmacokinetic (PK) and clinical efficacy of multiple doses of bimekizumab in patients with PsA (NCT02141763). The study was conducted in accordance with the applicable regulatory and International Council for Harmonisation, Good Clinical Practice requirements, the ethical principles originating in the Declaration of Helsinki and local laws. The protocol was reviewed and approved by the relevant ethics committees. All patients provided documented consent prior to study entry. The study was conducted at three sites, one each in the Republic of Moldova, the UK and Bulgaria, between June 2014 and August 2015.
Patients were randomised to one of five treatment regimens: intravenous administration on three separate occasions (weeks 0, 3 and 6) of one of the following dose regimens or matched placebo:
bimekizumab 240 mg/160 mg/160 mg (n=21)
bimekizumab 80 mg/40 mg/40 mg (n=6)
bimekizumab 160 mg/80 mg/80 mg (n=6)
bimekizumab 560 mg/320 mg/320 mg (n=6)
Concomitant medication was permitted, with adjustments allowed after week 8.
The cohort of patients receiving bimekizumab 240 mg/160 mg/160 mg or placebo (intravenous; n=3) was administered treatment first; the remaining three cohorts received bimekizumab or placebo (intravenous; n=3) in parallel following safety and PK reviews of the 240 mg/160 mg/160 mg cohort. Patients were randomised via interactive response technology; the randomisation schedule was produced by the clinical research organisation; no stratification factors were used in the process. Details on maintenance of study blind and changes to the planned study protocol are provided in the online supplementary information.
Patients
Key inclusion criteria: male or female, ≥18 years at screening; diagnosis of adult-onset PsA ≥6 months prior to screening, classified according to the Classification Criteria for PsA (CASPAR); active psoriatic lesions or history of skin PSO; active arthritis as defined by ≥3 tender joints and ≥3 swollen joints at screening and baseline, and either erythrocyte sedimentation rate of ≥28 mm/hour or high sensitivity C reactive protein >3 mg/L; inadequate response to ≥1 non-biological disease-modifying anti-rheumatic drug (DMARD, including methotrexate) and/or one approved biological DMARD; and receiving concurrent methotrexate for ≥3 months at the time of screening. Key exclusion criteria: inadequate response to >1 approved biological DMARD (no specific medication detailed); absolute neutrophil count <1.50×109/L and/or lymphocyte count <1.00×109/L.
Assessments
Primary safety endpoints assessed during the study included incidence and types of adverse events (AEs); clinical laboratory measurements and vital signs; echocardiogram; physical examination. Primary PK analyses included Cmax, CmaxSS, CminSS, area under the curve (AUC)tau, tmax, CL and V. Exploratory efficacy endpoints included American College of Rheumatology (ACR) scores; Psoriasis Area and Severity Index (PASI); physician’s global assessment of disease activity (PGA); patient’s global assessment of disease activity (PtGA, using a visual analogue scale). Efficacy endpoints were evaluated during screening, baseline and weeks 1 (PASI and PGA only), 2, 3, 6, 8, 12, 16 and 20. Blood samples for determining the plasma concentration of bimekizumab and anti-bimekizumab antibodies were taken throughout the study; concentration of anti-drug antibodies was an exploratory endpoint.
Statistical analysis
For the blockade experiments in synoviocytes and primary NHDFs, a one-way analysis of variance (ANOVA) with Fisher’s least significant difference (LSD) post-hoc test was used to assess overall differences between the groups and IgG control. A one-tailed paired t-test was chosen for the comparison between the anti-IL-17A and bimekizumab groups. Mean and standard error of the mean (SEM) were presented.
As predefined in the analysis plan, data from the three highest bimekizumab dose groups were pooled (the lowest dose group was expected to produce suboptimal efficacy and was therefore only included in the safety and PK data sets) and all placebo data were also pooled for summary and statistical inference purposes.
A sample size of 50 was deemed sufficient to meet the primary study objectives (safety, tolerability and PK), and to detect a difference of the pooled treatment group from pooled placebo group in ACRn with >90% probability based on a frequentist approach. In terms of ACR20, this power is based on an approximate target effect of 70% for bimekizumab and 25% for placebo.
The analyses of ACRn and ACR20 at week 8 followed the Bayesian paradigm to improve the operating characteristics of the study design. The Bayesian methodology was used for the analysis of ACRn and ACR20 at week 8 assuming informative priors only on the placebo response (vague prior distributions were assumed for all other parameters). For both endpoints, the prior placebo response, in terms of ACR20 response rate, was approximately 25% and the effective sample size was 32. Normal likelihood model and logistic model were assumed for ACRn and ACR20, respectively.
Two study efficacy criteria were predefined based on ACRn at week 8. For declaring PoC, we required a high (≥97.5%) posterior probability that bimekizumab is superior to placebo. To give further confidence about the effect size, the posterior probability that bimekizumab improvement over placebo exceeds a clinically relevant effect (ie, approximately 25% difference from placebo in ACR20 terms) was required to be ≥70%. The clinically relevant effect was determined after assessment of the efficacy data from a TNF inhibitor clinical study (NCT01087788).15 For ease of interpretation, posterior estimates and 95% credible intervals (CrI) for ACR20 only are presented.
Further details on the statistical methods are described in online supplementary information.
Results
Preclinical methods
IL-17F was expressed in disease-relevant cells and triggered a qualitatively similar inflammatory response to IL-17A
Several expression studies have established the presence of not only IL-17A but also IL-17F protein in lesional psoriatic skin.16 17 We detected both IL-17A and IL-17F in synovial tissue from patients with PsA using mRNA expression analysis, supporting a potential contribution of IL-17F to the immunopathology of PsA (online supplementary figure s1).
Because the receptor complex for IL-17A and IL-17F (IL-17RA and IL-17RC) is expressed in psoriatic skin16 and in synovitis,18–20 we next investigated whether IL-17F triggers a pro-inflammatory response in PsA synoviocytes and NHDFs. Whereas neither IL-17A nor IL-17F substantially induced activation of PsA synoviocytes by themselves, both IL-17 family members induced significantly greater production of key pro-inflammatory mediators such as IL-8 (figure 1A) and IL-6 (online supplementary figure s2) when synoviocytes were stimulated in the presence of TNF, with IL-17F being less potent than IL-17A. In primary NHDFs, amplification of IL-8 production was also observed following stimulation with recombinant IL-17A and IL-17F in the presence of TNF (figure 1B). Collectively, these data indicate that IL-17A and IL-17F are co-expressed in disease-relevant cells of patients with PsA and that, similar to IL-17A, IL-17F triggers a pro-inflammatory response in key effector cells from these tissues.

IL-17F contributes to inflammation and bimekizumab demonstrates superior efficacy relative to inhibition of IL-17A or IL-17F alone. Recombinant IL-17A and IL-17F, with or without TNF, were used to activate either (A) PsA synoviocytes (n=2) or (B) NHDFs (n=4), and IL-8 release was assessed following overnight culture. To evaluate the individual and collective influence of IL-17A and IL-17F, (C) PsA synoviocytes (n=4), (D, F, G) primary NHDFs (n=4) or (E) normal synoviocytes (n=5) were stimulated with Th17 supernatant with or without IL-17-specific blocking antibodies. Following overnight culture, either (C, D) inhibition of IL-8 production, (E, F) gene transcriptional changes or (G) inhibition of chemotactic potential was evaluated. For transcriptional analysis (E, F), genes were normalised to GAPDH mRNA and expressed as relative fold changes compared with unstimulated cells. For primary NHDFs, the panel presents genes that had a fold change ≥3 for Th17 stimulation under IgG conditions. Values were subject to conditional formatting (three colour scale: maximum value—red, minimum value—green, median value—yellow) for each target gene across groups. The same genes were analysed for normal synoviocytes and the conditional formatting was applied for genes expressing a fold change ≥3. Data are mean ±SEM. Figures are representative of three independent experiments. *represents a significant reduction versus IgG control, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. † represents a significant reduction versus anti-IL-17A, †P<0.05, ††P<0.01, ††††P<0.0001. IL, interleukin; NHDF, normal human dermal fibroblasts; PsA, psoriatic arthritis.
Dual neutralisation of IL-17A and IL-17F demonstrated greater suppression of synoviocyte and primary normal human dermal fibroblasts activation than blockade of IL-17A or IL-17F alone
To assess if IL-17F contributes to chronic tissue inflammation beyond IL-17A, synoviocytes from patients with PsA and primary NHDFs were stimulated with the supernatant from polyclonal human Th17 cells (online supplementary figure s3). Blockade of IL-17A alone significantly downregulated production by synoviocytes of pro-inflammatory mediators such as IL-8, whereas IL-17F blockade had no significant effect. Dual neutralisation of IL-17A and IL-17F by bimekizumab resulted in greater downregulation of IL-8 (28% lower; P<0.05) (figure 1C), IL-6 (42% lower; P<0.01) and MMP3 (44% lower; P=0.149) (online supplementary figure s4) than IL-17A blockade alone. Similar results were obtained when IL-17A and IL-17F were simultaneously neutralised by adding both single blocking antibodies, suggesting that the superior effect of bimekizumab was due to more than simply potent blockade of IL-17A (figure 1C). Moreover, blockade by an anti-TNF antibody had only a modest impact in this model system (figure 1C).
A similar analysis using primary NHDFs confirmed that dual neutralisation of IL-17A and IL-17F by bimekizumab resulted in a greater reduction in IL-8 (57% lower; P<0.0001) (figure 1D) and IL-6 (28% lower; P<0.01) (online supplementary figure s4), compared with IL-17A blockade alone; single IL-17F blockade had only a minimal effect.
Transcriptional analysis in Th17-stimulated normal synoviocytes and primary NHDFs (figure 1E, F) confirmed that dual neutralisation of IL-17A and IL-17F with bimekizumab resulted in a more profound anti-inflammatory effect in a large panel of inflammation-linked genes, versus inhibition of IL-17A alone. To confirm this concept in a functional assay, we investigated changes in the chemotactic potential of neutrophils towards Th17-stimulated NHDFs. While single cytokine neutralisation of IL-17A or IL-17F suppressed migration of neutrophils compared with the isotype control IgG (figure 1G), significantly more inhibition of neutrophil migration was achieved when both IL-17A and IL-17F were neutralised by bimekizumab (online supplementary video s1).
Supplementary file 2
Collectively, these in vitro data indicate, despite the inferior potency compared with IL-17A, that IL-17F has a role in driving inflammatory activation and that dual neutralisation of IL-17A and IL-17F more profoundly suppresses the inflammatory response of disease-relevant cells than blockade of IL-17A alone.
Clinical
Baseline disease characteristics, safety and pharmacokinetics
To assess the potential of dual IL-17A and IL-17F blockade in vivo, 53 of 80 patients screened for inclusion were randomised to treatment with bimekizumab (n=39), or placebo (n=14) for 20 weeks; 50 patients completed the study (online supplementary figure s5). Disease characteristics were representative for people with moderate-to-severe PsA (table 1). All patients received concomitant medication during the study; the most common were anti-inflammatory and anti-rheumatic products (100%), folic acid (59.6%) and analgesics (26.9%); 84.6% of patients received concomitant DMARDs.
Demographics and baseline characteristics (FAS)
Bimekizumab treatment was not associated with any unexpected safety signals; treatment-emergent adverse events (TEAEs), the majority of which were mild or moderate (13 (93%) patients in the placebo group and 36 (95%) patients in the total bimekizumab group), are summarised in table 2. Comparing across the individual bimekizumab groups, there was no apparent dose relationship with regard to the incidence of TEAEs. Three patients reported severe AEs (one ‘injuries caused by fall’ and one ‘muscle spasms’ in the bimekizumab treatment group; one ‘worsening of PsA’ in the placebo arm); no severe AEs were considered treatment related. One patient receiving bimekizumab reported three serious adverse events (SAEs) caused by a fall; none were considered treatment related. One oropharyngitis fungal infection and one vulvovaginal candida infection were reported in the bimekizumab arm (days 9 and 43 post infusion, respectively); both of these were mild, of short duration (≤20 days) and resolved with anti-fungal therapy. No patients discontinued because of TEAEs and there were no deaths. Mean plasma concentration–time profile data for bimekizumab are summarised in figure 2. Plasma concentrations of bimekizumab increased with dose in a linear and time-independent manner with linear clearance; the estimated t1/2 of bimekizumab was 24 days.
Safety outcomes (FAS)
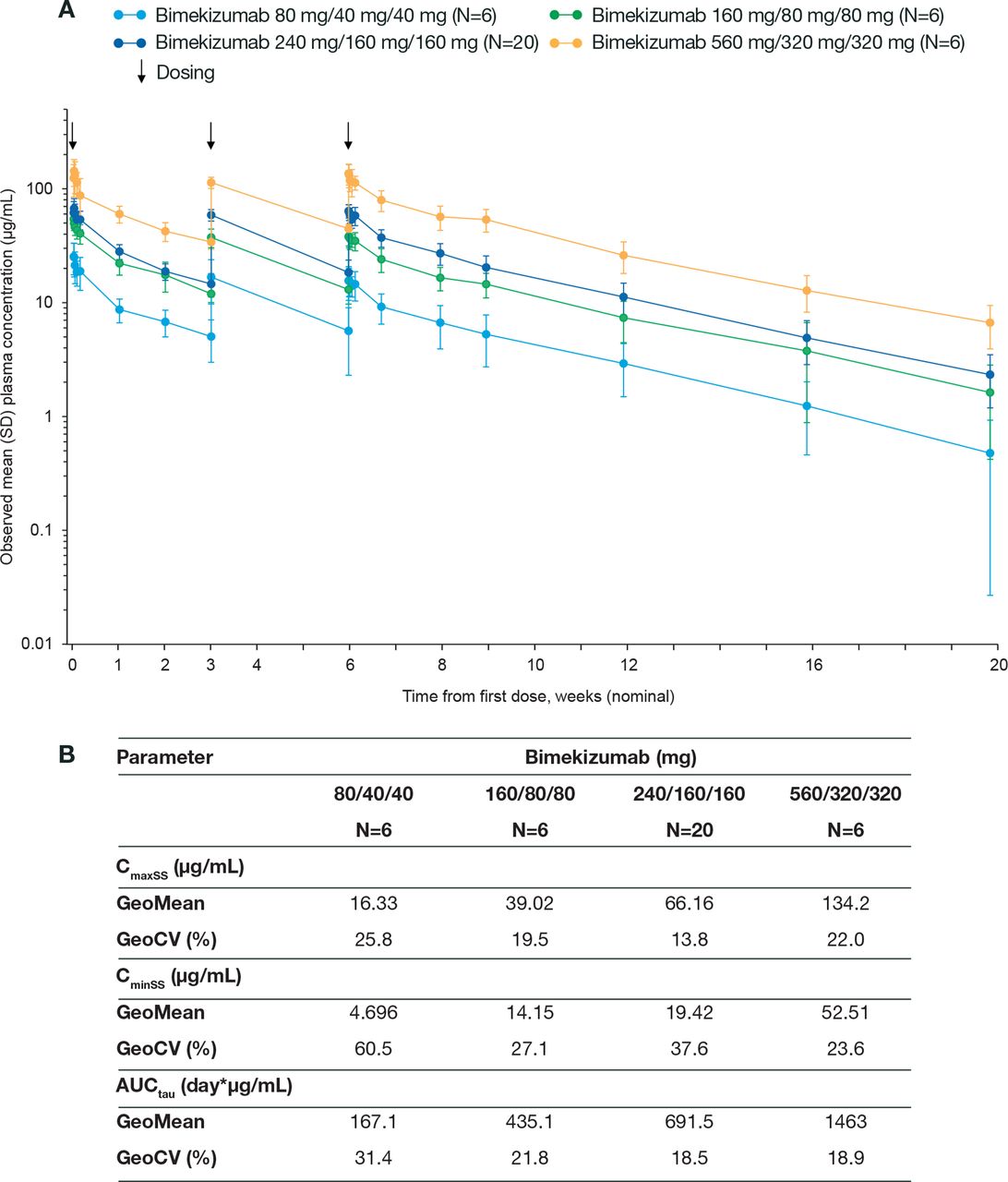
Pharmacokinetic parameters of bimekizumab (PK-PPS). (A) GeoMean plasma concentration–time profile of bimekizumab (PK-PPS) and (B) table of pharmacokinetic variables of bimekizumab. PK-PPS, pharmacokinetic per-protocol set. Note: Means, SDs and CVs were only calculated if at least one-third of the concentrations were quantified at a respective time point. CV, coefficient of variation.
Proof of concept: clinical efficacy of bimekizumab on joints and skin in patients with psoriatic arthritis
A summary of the clinical efficacy of the three highest dose groups of bimekizumab on measures of disease activity in joints and skin is presented in table 3. The prespecified efficacy success criteria of this study were met as Bayesian analysis indicated a >99% probability that joint response rates, as defined by ACRn and ACR20, at week 8 are greater with bimekizumab than with placebo (online supplementary table s2 and s3). Additionally, there was also a >99% probability that the ACRn and ACR20 response rates with bimekizumab at week 8 exceeded a clinically relevant threshold (defined based on results from a TNF inhibitor clinical study (NCT01087788)).15 Therefore, both of the prespecified efficacy success criteria of this study were met.
Clinical efficacy of bimekizumab on disease activity measures in joint and skin (PD-PPS)
Patients receiving bimekizumab had greater ACR20, ACR50 and ACR70 joint response rates compared with placebo (table 3). Response rates at the primary efficacy endpoint (week 8) were 80% (ACR20), 40% (ACR50) and 23% (ACR70); maximal observed response rates for these variables were 80% (ACR20 at week 8), 57% (ACR50 at week 12) and 37% (ACR70 at week 16) (figures 3A, B and C). A response was evident as early as week 2 and maximal or near-maximal responses were maintained to week 20.

{kind=link}
{kind=link}
{kind=link}
ACR and PASI response rates by treatment group (PD-PPS). (A–C) ACR response rates (%, with 95% CI) and (D-E) PASI response rates (%, with 95% CI). Dashed vertical lines indicate drug intake (weeks 0, 3 and 6). Note: top three doses of bimekizumab (160 mg/80 mg/80 mg, 240 mg/160 mg/160 mg, 560 mg/320 mg/320 mg); PASI75 and PASI100 were only calculated for patients with BSA ≥3% psoriasis involvement at baseline. Modifications in concomitant therapy were permitted post week 8. Subject 003–00320 was excluded from the analyses for all time points after week 8 due to receiving an increased dose of naproxen, adalimumab and leflunomide for control of worsening disease. ACR20, American College of Rheumatology 20% response criteria; ACR50, American College of Rheumatology 50% response criteria; ACR70, American College of Rheumatology 70% response criteria; PASI75, Psoriasis Area and Severity Index 75% response criteria; PASI100, Psoriasis Area and Severity Index 100% response criteria; PD-PPS, pharmacodynamic per-protocol set.
In patients with significant skin involvement (BSA≥3%), week 8 response rates for PASI75 and PASI100 were 100% and 87%, respectively. As with the joint outcomes, there was a response as early as week 2, and maximal or near-maximal responses were maintained to week 20 (table 3; figure 3D and E).
PtGA and PGA demonstrated improvements from baseline within 2 weeks of the first infusion for patients treated with bimekizumab versus placebo (online supplementary figure s6; table 3).
Discussion
A combined preclinical and clinical PoC approach was used to probe the hypothesis that not only IL-17A, but also IL-17F, contributes to chronic tissue inflammation, and that dual neutralisation of IL-17A and IL-17F by bimekizumab could profoundly suppress joint and skin inflammation in PsA. The rapid and marked improvements in joint and skin outcomes seen in this PoC study not only exceeded placebo but were superior in joints to a clinically relevant effect (based on the effect of current standard of care (SoC)).
Our preclinical data indicate that IL-17A and IL-17F are not only co-expressed in PsA target lesions, consistent with a recent report in the literature,21 but that IL-17F has a qualitatively similar pro-inflammatory effect to IL-17A on disease-relevant human tissue cells. Crucially, neither IL-17A nor IL-17F are potent inflammatory cytokines on their own, but rather co-operate with other inflammatory mediators, including, but not restricted to, TNF.22 23 In addition, IL-17A was more potent than IL-17F in the activation of human synoviocytes and primary NHDFs; however, the implications of this should be considered in the context of observations that levels of IL-17F protein are 32-fold higher than IL-17A in lesional psoriatic skin (317 vs 9.8 pg/mL).24 Collectively, these data suggest that while IL-17F may not play a dominant inflammatory role in the presence of high IL-17A levels, IL-17F may become relevant in complex cytokine micro-environments.
Supporting the hypothesis that IL-17F contributes to chronic tissue inflammation beyond IL-17A alone, we demonstrated that, in disease-relevant human cellular systems, dual neutralisation of IL-17A and IL-17F had a more profound impact on the inflammatory activation of synoviocytes and primary NHDFs by Th17 cell supernatant than selective blockade of IL-17A alone. This was observed not only in terms of gene and protein expression but also translated to functional consequences, such as impact on neutrophil migration.
To examine the hypothesis that neutralisation of both IL-17A and IL-17F would lead to superior inhibition of inflammatory responses than blockade of IL-17A alone, the impact of cytokine-specific antibodies on disease-relevant tissues stimulated with Th17-supernatant was assessed. As predicted, neutralisation of IL-17F when in the presence of sufficient quantities of IL-17A resulted in only modest inhibition of the inflammatory response, whereas IL-17A inhibition showed a marked effect. However, dual neutralisation of IL-17A and IL-17F with bimekizumab demonstrated superior neutralisation versus inhibition of IL-17A alone. Use of an affinity-matched anti-IL-17A combined with an anti-IL-17F selective antibody confirmed that the anti-inflammatory effects of bimekizumab were due in part to the anti-IL-17F component and not simply a result of superior IL-17A blockade. The limited effect of IL-17F in the presence of IL-17A may explain why its pro-inflammatory role has been overlooked previously.
Because our recent single-ascending-dose phase I trial with bimekizumab in patients with mild PSO revealed no unexpected safety signals, and provided preliminary evidence of a profound effect on reduction of psoriatic lesions,25 we proceeded to assess the potential of bimekizumab in vivo in patients with PsA by conducting a PoC clinical trial. The trial did not reveal new safety signals; the majority of TEAEs, including neutropenia and liver transaminase elevations, were mild to moderate in intensity and resolved spontaneously. There were no cases of death, malignancy or major cardiovascular events. There were three severe AEs and three SAEs, none of which were considered related to the treatment.
In the context of previous reports with anti-IL-17A and anti-IL-17RA antibodies,26–28 it is important to note the absence of opportunistic infections (including tuberculosis), inflammatory bowel disease and suicide-related events in this trial. In line with the increased susceptibility for fungal infections emerging with anti-IL-17A antibodies, we observed two cases of candidiasis (oropharyngeal and vulvovaginal), both of which were mild and resolved with local treatment.
The clinical study met its predefined efficacy criterion, demonstrating superiority of bimekizumab over both placebo and the predefined clinically relevant threshold using ACRn at week 8. Currently, anti-TNF therapy is considered the SoC biological treatment for PsA; therefore, the clinically relevant threshold was based on a pivotal randomised controlled trial with TNF blockade in patients with PsA (NCT01087788).15
Studies of other biologics targeting the IL-17 pathway have demonstrated efficacy in PsA.26 28 29 ACR20 response rates for secukinumab (anti-IL-17A mAb) were 54% (300 mg subcutaneous) at week 24 versus 15% for placebo.26 A phase 3 study of ixekizumab (anti-IL-17A mAb) demonstrated an ACR20 response of 62% (80 mg subcutaneous Q2W) at week 24 versus 30% for placebo.28 The ACR20 response rate for brodalumab (anti-IL-17RA mAb) was 39% at week 12 (280 mg subcutaneous) versus 18% for placebo.30 While the size and design of the current trial precludes any direct or indirect comparison between these published trials and SoC, the superiority of bimekizumab over the predefined clinically relevant threshold and the observed ACR20 responses (reaching 80% vs 17% for placebo at week 8) are encouraging. Alongside this, ACR50 (reaching 57% at week 12), ACR70 (reaching 37% at week 16), strong skin responses (PASI75 and PASI100 of 100% and 87%, respectively, at week 8) and rapid onset of action (some responses observed within 2 weeks of first infusion) concord to indicate robust therapeutic efficacy of bimekizumab within the contextual limitations of a PoC trial.
When evaluating these data, the size and duration of the PoC trial limits the interpretation of the safety dataset; furthermore, the small sample size warrants cautious interpretation of the observed therapeutic effect size. Because patients received only three doses of bimekizumab, further optimisation of the dosing regimen may impact the onset, level and durability of clinical responses in joint and skin. Of note, in the current trial, modifications of background medication were allowed after week 8, potentially explaining the increase in ACR responses seen in the placebo group between weeks 8 and 20. Imbalances in distribution of concomitant medications observed across treatment groups may also have impacted our findings, and outcomes should be interpreted with caution due to the small sample size.
In summary, the study supports our hypothesis that IL-17F plays a role in chronic tissue inflammation beyond that of IL-17A, and that dual neutralisation of IL-17F and IL-17A has a clinical impact on patients. The rapid, profound, and sustained joint and skin responses in the PoC trial with bimekizumab in active PsA support further clinical investigation of dual inhibition of IL-17A and IL-17F in immune-mediated inflammatory diseases.
Acknowledgments
The authors acknowledge the contributions of Shauna West, of UCB, to the bimekizumab affinity analysis work, and Sophie Archer PhD, Tim Smallie, of UCB, Louise Healy, formerly of UCB, for their work on functional characterisation of bimekizumab. The authors also acknowledge the contribution of Catherine Simpson, of UCB, for flow cytometry cell sorting support; Remi Okoye, of UCB, for the immunohistochemistry work; and Iris Blijdorp of the Academic Medical Center, Amsterdam, for work on blockade experiments in synoviocytes. Additional statistical support was provided by Emma Jones of Veramed and Ros Walley of UCB. The authors would like to acknowledge Ailsa Dermody, PhD of iMed Comms, an Ashfield Company, part of UDG Healthcare plc, for medical writing support that was funded by UCB Pharma in accordance with Good Publication Practice (GPP3) guidelines. The authors acknowledge Alvaro Arjona, PhD of UCB Pharma, for publication and editorial support. The authors thank the patients and their caregivers, in addition to the investigators and their teams, who contributed to this study.
References
Footnotes
SG and DB are co-first authors.
Handling editor Tore K Kvien
Contributors Study design and/or interpretation of results: DB, TB, MG, SG, ADGL, AM, RO, FS, SS, PV, MILW, NY; acquisition and/or analysis of data: TB, MG, AM, LI, ADGL, PM, SP, FS, SS, PV, MILW, NY; drafting and/or revising manuscript: DB, TB, MG, SG, LI, ADGL, AM, PM, RO, SP, FS, SS, PV, MILW; specialised role in the research: TB (antibody design and characterisation), ADGL (BKZ rational design), SS (IL-17 biology expert), FS (statistical oversight); administrative, technical or supervisory support: SG, MG, AL. UCB Pharma funded the study and the development of the manuscript, and reviewed for scientific accuracy.
Funding The study was supported by UCB Pharma.
Competing interests DB, TB, MG, SG, LI, ADGL, AM, RO, SS, FS, PV, MILW are employees of UCB Pharma. DB, TB, ADGL, PV hold stocks and/or stock options in UCB Pharma. DB is a part-time employee of UCB Pharma and holds a part-time position at the Academic Medical Center/University of Amsterdam. DB received a grant from UCB Pharma to conduct preclinical experiments; DB received grants and/or consultant or investigator fees from the following organizations outside of the submitted work: AbbVie, Pfizer, MSD, Roche, BMS, Novartis, Eli Lilly, Boehringer Ingelheim and Glenmark. MG is a paid contractor for UCB working in a consulting capacity. PM is a scientific advisor to UCB Pharma and received associated fees outside of the submitted work. SP, NY declare no relevant conflicts of interest.
Ethics approval Ethics Committee at MC Comac Medical; National Ethics Committee; GM South REC.
Provenance and peer review Not commissioned; externally peer reviewed.