Article Text

Abstract
Familial cold autoinflammatory syndrome, Muckle-Wells syndrome (MWS), and chronic, infantile, neurological, cutaneous and articular (CINCA) syndrome are dominantly inherited autoinflammatory diseases associated to gain-of-function NLRP3 mutations and included in the cryopyrin-associated periodic syndromes (CAPS). A variable degree of somatic NLRP3 mosaicism has been detected in ≈35% of patients with CINCA. However, no data are currently available regarding the relevance of this mechanism in other CAPS phenotypes.
Objective To evaluate somatic NLRP3 mosaicism as the disease-causing mechanism in patients with clinical CAPS phenotypes other than CINCA and NLRP3 mutation-negative.
Methods NLRP3 analyses were performed by Sanger sequencing and by massively parallel sequencing. Apoptosis-associated Speck-like protein containing a CARD (ASC)-dependent nuclear factor kappa-light chain-enhancer of activated B cells (NF-κB) activation and transfection-induced THP-1 cell death assays determined the functional consequences of the detected variants.
Results A variable degree (5.5–34.9%) of somatic NLRP3 mosaicism was detected in 12.5% of enrolled patients, all of them with a MWS phenotype. Six different missense variants, three novel (p.D303A, p.K355T and p.L411F), were identified. Bioinformatics and functional analyses confirmed that they were disease-causing, gain-of-function NLRP3 mutations. All patients treated with anti-interleukin1 drugs showed long-lasting positive responses.
Conclusions We herein show somatic NLRP3 mosaicism underlying MWS, probably representing a shared genetic mechanism in CAPS not restricted to CINCA syndrome. The data here described allowed definitive diagnoses of these patients, which had serious implications for gaining access to anti-interleukin 1 treatments under legal indication and for genetic counselling. The detection of somatic mosaicism is difficult when using conventional methods. Potential candidates should benefit from the use of modern genetic tools.
- CAPS
- NLRP3
- somatic mosaicism
- massively parallel sequencing
Statistics from Altmetric.com
Cryopyrin-associated periodic syndromes (CAPS) are a group of autoinflammatory diseases that include familial cold autoinflammatory syndrome, Muckle-Wells syndrome (MWS), and chronic, infantile, neurological, cutaneous and articular (CINCA) syndrome, also known as neonatal-onset multisystem inflammatory disease (NOMID).1 Some clinical features are shared by almost all CAPS phenotypes (ie, onset during childhood, an urticaria-like skin rash) whereas others are restricted to certain phenotypes (ie, serum amyloid A protein (AA) amyloidosis in MWS, destructive arthropathy in CINCA-NOMID).1 CAPS are caused by dominantly inherited or de novo NLRP3 mutations.2–4 This gene encodes for cryopyrin, a component of one of the cytosolic complexes named inflammasomes that generate the active form of interleukin 1ß (IL-1ß).5 Previous studies showed a gain-of-function behaviour for those NLRP3 mutations associated with CAPS because they provoke an uncontrolled IL-1ß overproduction, representing the basis from which to treat these patients with anti-IL-1 drugs.3 ,6 Genetic heterogeneity was suggested in CINCA-NOMID because only ≈55% of patients was NLRP3 mutation-positive.3 ,4 The use of novel genetic methods recently detected somatic NLRP3 mosaicism in ≈35% of patients with CINCA-NOMID.7 ,8 However, no data are currently available about the role of this genetic mechanism in other CAPS phenotypes because genetic heterogeneity has hitherto been scarcely reported in previous studies.
We herein show the causal role of somatic NLRP3 mosaicism in patients with MWS, in whom previous studies did not detect NLRP3 mutations, suggesting that this genetic mechanism is shared among the different CAPS phenotypes.
Patients and methods
Patients
For this study we enrolled patients with a clinical suspicion of CAPS, with a phenotype of MWS and overlapping syndromes, and NLRP3 mutation-negative in previous studies. The clinical inclusion criteria were the presence of an urticaria-like skin rash and at least one of the following symptoms: recurrent fever, recurrent arthritis, recurrent aseptic meningitis, sensorineural deafness or AA amyloidosis (see online supplementary table S1 for details). All patients with a CINCA-NOMID phenotype were excluded. The patients’ data were collected by direct interviews and chart reviews. Written informed consent from patients (or patients’ parents if younger than 18-years-old) was obtained at each institution. The ethics committees of Hospital Clinic, Barcelona and the Graduate School of Medicine, Kyoto University approved this study, which was conducted in accordance with the Helsinki Declaration.
NLRP3 analyses
These analyses were performed in the Graduate School of Medicine, Kyoto University or in the Hospital Clínic, Barcelona. Genomic DNA was obtained from whole peripheral blood using QIAmp DNA Blood Mini Kit (QIAgen, Germany). For Sanger sequencing all exons of NLRP3 gene were amplified by PCR using the primers and conditions previously described.2 The PCR amplicons were purified with Illustra ExoStar 1-Step kit (GE Healthcare, USA), bidirectional fluorescence sequencing using ABI BigDye Terminator V.3.1 Cycle Sequencing Kit (Applied Biosystems, USA) and run on an automated ABI 3730XL DNA analyzer. For massively parallel DNA sequencing, all exons of NLRP3 gene were amplified as previously described.8 Library preparation and emulsion PCR were performed according to manufacturer's instructions. All sequencing runs were performed on the GS Junior 454 Sequencer using the GS Junior Titanium Sequencing kits (Roche, Switzerland). The obtained sequences were analysed using the Amplicon Variant Analyzer software.
Bioinformatics analyses
In silico sequence analyses were performed using two different algorithms. The Sorting Intolerant from Tolerant is a sequence homology based tool that predicts whether the amino acid substitution is or is not probably damaging by reporting a score. The PolyPhen-2 is a tool for prediction of the possible impact of an amino acid substitution on the structure and function of a protein, and qualitatively appraised as benign, possibly damaging or probably damaging.9 ,10
Functional studies
The functional consequences of the novel NLRP3 variants were evaluated in two in vitro assays.11 Wild type and mutant NLRP3 cDNA, obtained by mutagenesis PCR, were subcloned into the expression vectors pEF-BOSEX and pcDNA5/TO (Invitrogen, USA). The Apoptosis-associated Speck-like protein containing a CARD (ASC)-dependent nuclear factor kappa-light chain-enhancer of activated B cells (NF-κB) activation was evaluated using a dual-luciferase reporter assay in HEK293FT cells transfected with NLRP3-pEF-BOSEX plasmids with a NF-kB reporter construct (pNF-kB-luc, BD Biosciences) and an internal control construct (pRLTK, Toyo Ink) in the presence or absence of ASC-expression plasmid. To evaluate the necrosis-like cell death, the THP-1 cell line (a human monocytic cell line derived from a patient with acute monocytic leukemia) was transfected with green fluorescent protein (GFP)-tagged NLRP3-pcDNA5/TO plasmids. After 4 h, cells were stained with 7-aminoactinomycin D and cell death of GFP positive cell was analysed by FACS Caliber (Becton-Dickinson).
Statistical analyses
Continuous variables are presented as the mean±SD or as the median and IQR, while categorical variables are presented as numbers, ratios and/or percentages. To detect potential differences among patients with germline mutations and with somatic mutations, the Mann-Whitney U test was used for continuous variables and Fisher's exact test was used for categorical variables.
Results
Genetic analyses
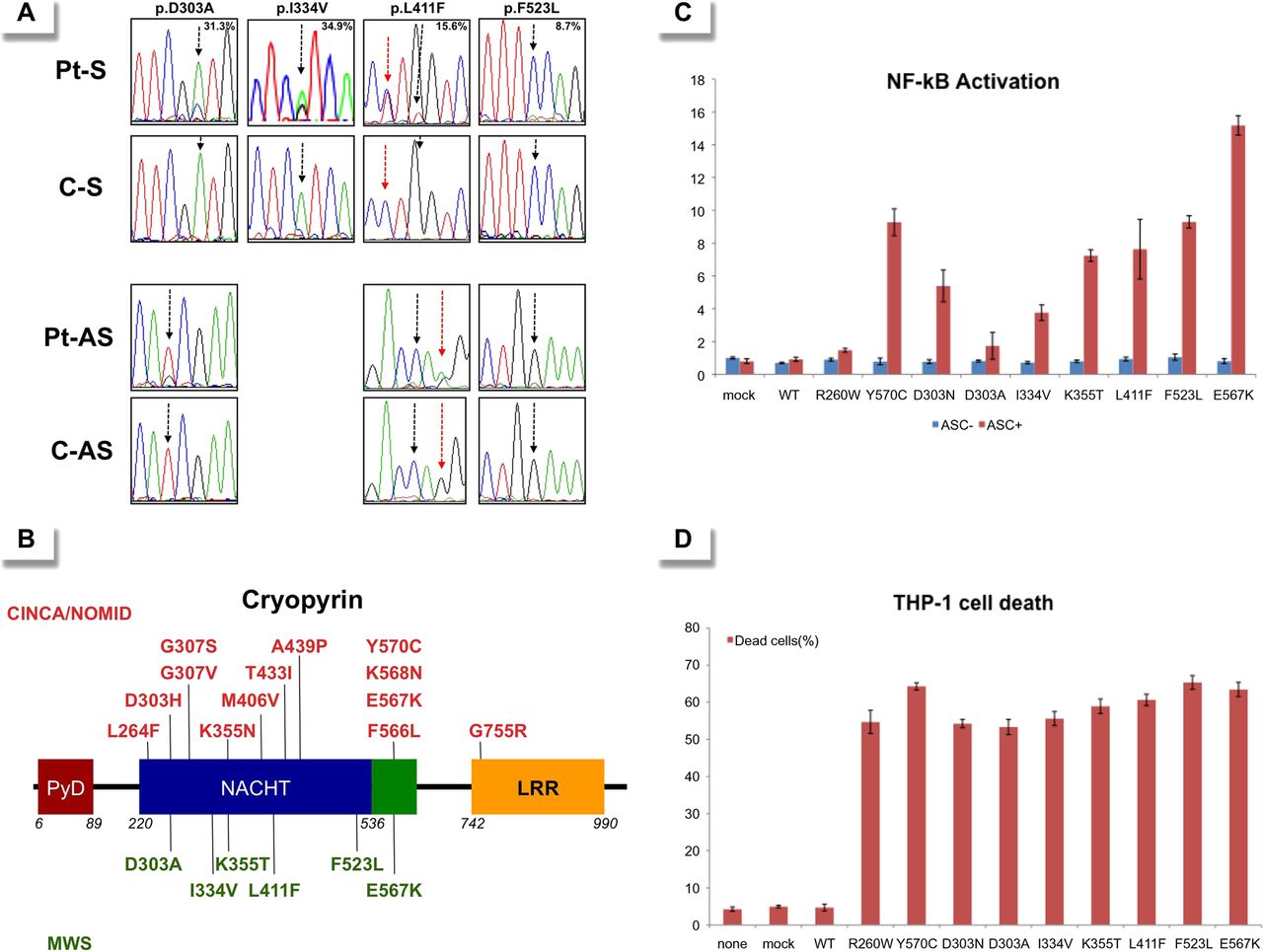
Fifty-six patients (23 Japanese and 33 Spanish) who fulfilled the inclusion criteria were enrolled. Sanger sequencing of the NLRP3 gene did not identify mutations in any patients. However, small peaks with reduced signal intensities compared with controls were detected in two patients: the A-to-C transversion at c.908 position in Patient 1 and the A-to-G transition at c.1000 position in Patient 2, which encode for the p.Asp303Ala and p.Ile334Val cryopyrin variants, respectively (figure 1A and table 1). Massively parallel DNA sequencing was performed in all patients and revealed somatic NLRP3 mosaicism in seven patients (7/56; 12.5%). Six different nucleotide changes, all of them located in the exon 3, were detected, and their frequency varied notably among patients, ranging from 5.5% to 34.9% (table 1). All NLRP3 variants encode for non-synonymous amino acid changes, three of them being novel (p.Asp303Ala, p.Lys355Thr and p.Leu411Phe) and the remainder already described (p.Ile334Val, p.Phe523Leu and p.Glu567Lys) (figure 1B). In Patient 4 the frequency of the mutated NLRP3 allele remained identical in blood samples obtained over an 8-year period (table 1).

(A) Sense (upper rows) and antisense (bottom rows) chromatograms from four patients with somatic NLRP3 mosaicism and controls obtained by Sanger sequencing using genomic DNA extracted from whole blood. The black arrows show the NLRP3 positions where the somatic mutations were detected. The percentage in the upper panels represents the frequency of the mosaicism obtained by massively parallel DNA sequencing in each patient. The red arrows indicate the c.1231 C>T NLRP3 polymorphism (rs#148478875). (B) Structural organisation of cryopyrin. Above the protein structure are indicated all missense cryopyrin variants that have been detected as somatic mutations in patients with chronic, infantile, neurological, cutaneous and articular (CINCA)-neonatal-onset multisystem inflammatory disease (NOMID) in previous reports, and those below the protein structure are the missense variants detected as somatic mutations in the present study. (C) ASC-dependent NF-kB activation and (D) necrotic THP-1 cell death, induced by the detected NLRP3 mutations. Values are the mean±SD of triplicate experiments, and data are representative of two independent experiments. AS, antisense; ASC, Apoptosis-associated Speck-like protein containing a CARD; C, control; LRR, leucine-rich repeat; mock, vector without NLRP3; MWS, Muckle-Wells syndrome; NACHT, a family of NTPases that originally included the NAIP, CIITA, HETE-E and TP-1 proteins; NF-kB, nuclear factor kappa-light chain-enhancer of activated B cells; None, nothing transfected; Pt, patient; PyD, pyrin domain; S, sense; WT, wild type NLRP3.
Summary of genetic data of patients with somatic NLRP3 mosaicism
Bioinformatics and functional analyses
All missense NLRP3 variants were predicted to be possibly or probably damaging to cryopyrin structure and/or function according to at least one of the two algorithms employed, with the only exception of p.Glu567Lys variant (table 1). Interestingly, this NLRP3 variant was twice detected in the unrelated patients with somatic mosaicism, and has also been reported in other patients with CAPS, reasonably supporting its pathogenic effect.7 ,11 We did not find any of the detected NLRP3 variants in two groups of ethnically matched healthy individuals (Japanese controls n: 200 chromosomes; Spanish controls n: 500 chromosomes) nor in the database National Center for Biotechnology Information (NCBI) single nucleotide polymorphism database (dbSNP) Build 137 (table 1), reasonably ruling out that they could be rare gene polymorphisms.
Finally we evaluated their functional consequences by two different in vitro assays. The results showed that all NLRP3 variants induced ASC-dependent NF-κB activation (figure 1C) and necrosis-like programmed cell death of THP-1 cell line (figure 1D) at a similar or higher level than those induced by other well-known disease-causing mutations (p.Arg260Trp, p.Asp303Asn and p.Tyr570Cys). Altogether, these data clearly support a pathogenic effect for all NLRP3 mutations detected as somatic mutations in the enrolled patients.
Clinical features of patients with somatic NLRP3 mosaicism
At the time of inclusion in the study, the clinical diagnosis of patients with somatic NLRP3 mosaicism was compatible with MWS. Neither consanguinity nor familial history of the disease was reported in any of them. The inflammatory disease started during their infancy or childhood (median: 4 years; IQR: 1.3–9.0 years), with an urticaria-like skin rash and a marked inflammatory acute response as the main features at that time (see table 2 for clinical details at the disease onset).
Summary of clinical features of patients with somatic NLRP3 mosaicism at the onset of the disease
All patients referred to the chronic course of their disease, with variable disease evolution (median: 20 years; IQR: 12–26 years). During this time, recurrent arthritis (6/7; 85.7%), headache (5/7; 71.4%) and recurrent conjunctivitis (4/7; 57.1%) mainly added to those features detected at the disease onset. None of these patients developed AA amyloidosis, whereas five of them (71.4%) developed progressive bilateral sensorineural deafness (see table 3 for a detailed summary of clinical features detected during the course of the disease).
Summary of clinical manifestations detected in patients with somatic NLRP3 mosaicism during the course of the disease
Outcome of anti-IL-1 blockade
Five patients with somatic NLRP3 mosaicism were treated with anti-IL-1 drugs. Only Patient 5 was treated with anakinra (100 mg/24 h subcutaneous for a duration of 20 months). Three patients only received canakinumab: Patient 2 (150 mg/8 weeks subcutaneous for a duration of 13 months), Patient 3 (2 mg/kg/8 weeks subcutaneous for a duration of 16 months) and Patient 6 (initial dose of 150 mg/4 weeks, subsequently increased up to 300 mg/4 weeks, for a duration of 14 months). Patient 7 was first treated with anakinra (1 mg/kg/24 h subcutaneous for a duration of 24 months) and subsequently switched to canakinumab (150 mg/8 weeks subcutaneous for a duration of 14 months). All patients showed a marked and sustained improvement while treated with anti-IL-1 drugs, with a complete remission of urticaria-like skin rash (5/5), fever (3/3), conjunctivitis (2/2) and aseptic meningitis (1/1), and marked benefits for arthritis (complete response in 75%) and headache (complete response in 75%, and marked improvement in 25%). Inversely, IL-1 blockade did not improve the sensorineural deafness (0/4). The clinical improvement was associated with sustained reductions of erythrocyte sedimentation rate and C reactive protein level, and normalisation of white blood cell, neutrophil and platelets counts, and haemoglobin level (see figure 2 for details).

{kind=link}
{kind=link}
Laboratory values obtained in the five patients treated with different anti-interleukin 1 drugs. Patient's graphics were ordered as follows: First, those graphics from the patient who only received treatment with anakinra (Pt 5), followed by those from patients who only received treatment with canakinumab (Pt 2, 3 and 6) and finally those from the patient who received both treatments (Pt 7). Vertical bars represent the mean±SD of values obtained during treatment periods. Horizontal discontinued lines represent the upper limit of the normal range, with the only exception of the haemoglobin box, in which this line represents the lower limit of the normal range. CRP, C reactive protein; ESR, erythrocyte sedimentation rate; PMN, polymorphonuclears; WBC, white blood cell count.
Comparative phenotype analyses
To identify potential clinical differences among patients with germline or with somatic NLRP3 mutations two cohorts of patients with MWS were compared. The group of patients with MWS with somatic NLRP3 mosaicism included the seven patients described here whereas the cohort of patients with MWS with germline mutations included 41 patients (13 Japanese and 28 Spanish) from our databases. In this last group the germline status was established by means of pedigree analyses and/or by massively parallel sequencing. As expected, the familial history of the disease was a significant variable between the two groups. No significant differences were detected among the main clinical features (fever, urticaria-like rash, joint, neurological and ocular involvements, and deafness) despite their variable frequency in each group (see table 4 for details). However, patients with somatic NLRP3 mosaicism seemed to have late onsets of the disease and of the sensorineural deafness, an increased incidence of arthritis and a reduced risk of developing AA amyloidosis, when compared with patients with germline mutations.
Comparison of main clinical data of patients carrying germline versus somatic NLRP3 mutations
Discussion
CINCA-NOMID syndrome represents the severest CAPS phenotype, and is usually a consequence of de novo NLRP3 mutations. Recent works have established its genetic basis, with ≈55% of patients carrying germline NLRP3 mutations and ≈35% carrying somatic NLRP3 mosaicism.3–4 ,7 ,11–16 However, no studies addressing the presence of somatic NLRP3 mosaicism have been undertaken in other CAPS phenotypes because genetic heterogeneity has been poorly described in them, with only five reported patients with NLRP3 mutation-negative MWS.17–19 This scenario prompted us to hypothesise that somatic NLRP3 mosaicism might be an underlying genetic mechanism in patients with other CAPS phenotypes. For this proposal two ethnically different cohorts of candidates were screened, and 12.5% of them (7/56) carried variable degree of somatic NLRP3 mosaicism in peripheral blood. Additional evidences, as shown here, definitively support that the detected NLRP3 variants are pathogenic and include their absence in panels of ethnically matched controls and in a database of genomic diversity, in silico analyses that predict their damaging effect for the function and/or structure of cryopyrin, and in vitro functional studies that clearly showed its gain-of-function behaviour. Taken together these evidences support that somatic NLRP3 mosaicism is a genetic mechanism shared by different CAPS phenotypes, and it is not restricted to CINCA-NOMID syndrome.
Among NLRP3 mutations detected 50% (3/6) were novel, representing an unexpected high proportion for a small cohort. Taking into account their consequences on the cryopyrin function it is conceivable to hypothesise that, in germline status, they could be incompatible with life. We have also found a marked variability in the degree of somatic mosaicism among patients, which may have important consequences. For diagnostic purposes the level of somatic mosaicism could be the determining factor in achieving a definitive genetic diagnosis. Those patients with mosaicism around, or higher than, 15% will probably be detected in conventional studies using Sanger's method by means of careful analyses, as we have shown in the patients’ chromatograms. However, those patients with frequencies of less than 15% are probably missed by Sanger sequencing and will only be detected by using new technologies that are not currently widely available. The differences of disease severity observed among patients with somatic mosaicism, including those from this study and those from previous reports, could be explained by different and cumulative factors, which probably cannot be independently analysed. These factors might include, at least, the type of amino acid exchange, its location in the cryopyrin, its functional consequence in the normal cryopyrin function, and the degree and tissue distribution of somatic mosaicism. We must also note that all known somatic NLRP3 mutations seem to be located in some few amino acid residues (303, 355, 567) or in small regions of cryopyrin (303–307, 433–439 and 566–570), probably representing hot spots for these types of mutations. Consequently these regions should be carefully analysed when using Sanger sequencing to identify potential carriers of somatic mosaicism.
All patients with somatic NLRP3 mosaicism were sporadic patients, with no affected relatives, which is notably different from patients with germline mutations (positive familial history in 65.9%). Their main clinical features were compatible with a MWS phenotype and similar to those previously described in patients with germline mutations, with the potential exceptions of a reduced incidence of AA amyloidosis, an increased incidence of recurrent arthritis, and slightly older ages at the disease onset and also at onset of sensorineural deafness. It is interesting to note that most patients (4/7; 57.1%) were misdiagnosed as having juvenile idiopathic arthritis when the disease started, a similar misdiagnosis previously reported in different inherited autoinflammatory diseases.20–23 Despite the evidence shown here, the actual frequency of somatic NLRP3 mosaicism is unknown and probably underestimated. In our study a potential bias in the selection of patients could exist because they were selected on the basis of the presence of an urticaria-like skin rash associated with other symptoms. Recent studies have described atypical CAPS presentations in patients with germline NLRP3 mutations in whom urticaria-like skin rash was nearly absent.24 ,25 These data suggest that clinical diversity of CAPS is probably wider than previously described and further studies are necessary to delineate the profile of potential candidates to carry somatic NLRP3 mosaicism.
The evidence obtained may have serious implications for patients, especially with regards to treatment and genetic counselling. The outcome of IL-1 blockade in patients with somatic NLRP3 mosaicism was nearly identical to those reported in patients with germline mutations.26 ,27 The only symptom that did not improve with IL-1 blockade was the sensorineural deafness. In this regard, apparently contradictory responses have been reported, with improvement or amelioration in some patients and no response in others.14 ,17 ,28–30 It has been suggested that the time of evolution of deafness previous to starting anti-IL-1 drugs could be a determining factor for the type of response, but probably additional and unknown factors could also play a role in this particular manifestation. We have also observed a notable delay in gaining access to anti-IL-1 drugs with respect to the disease onset (median: 20 years; IQR: 12–26 years), because these treatments were administered under legal indication once the definitive CAPS diagnosis was established by means of the identification of somatic NLRP3 mosaicism. Taking into account the excellent response observed to IL-1 blockade, it is reasonable to hypothesise that if this was started earlier it should have provoked the non-appearance of some severe complications such as deafness.
For an appropriate genetic counselling the scenario is extremely different in patients with CAPS with germline or with somatic mutations. In the case of germline mutations, the risk of transmission to future pregnancies is 50%. Inversely, the prediction of the risk of transmission in cases of somatic mosaicism is more complex, because it may vary in the different tissues, it is not usually determined in gonadal tissues, and its detection probably requires new sensitive genetic methods that are not widely available. The vertical transmission of a somatic mutation is an extremely rare event, with only one case recently described in MWS.31 Consequently, this possibility should be considered during the genetic counselling of these patients, although one of the main messages to patients is that its probability remains low.
We show that somatic NLRP3 mosaicism underlies MWS and is probably a shared genetic mechanism in different CAPS phenotypes, and not restricted to CINCA/NOMID syndrome. Its detection was achieved by using massively parallel sequencing, and functional studies confirmed the gain-of-function behaviour of the detected variants. The detection of somatic mosaicism has had serious clinical implications for patients, including access to treatment under legal indication, adequate follow-up and ensuring appropriate genetic counselling. Further studies are necessary to delineate the clinical phenotype of candidates to looking for somatic mosaicism, in which new sensitive genetic technologies should be used.
Acknowledgments
The authors thank the patients and their families for their participation in this study.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Handling editor Tore K Kvien
-
KN, EG-R, RN and JIA contributed equally.
-
Contributors KN, TH, JY, RN and JIA designed research, discussed data and wrote the paper. EG-R, ER-O, FR, EI, TY, KI, TK and OO performed genetic and functional investigations, discussed data and reviewed the manuscript. AS, TK, HU, JMC, JC, ST, NK, JLC-R, NO-C, JA, SJ-T, CV, JF-M, IC, JH-R, MM, MTD, MB, SB, MY, TK, RK, NA, KS, NI, MKS and NK provided clinical data and blood samples, discussed data and reviewed the manuscript.
-
Funding Supported by the Spanish Ministry of Health (FIS PS09/01182), by the Japan's Ministry of Health, Labor and Welfare, and by the Japan's Ministry of Education, Culture, Sports, Science and Technology.
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval The ethics committees of Hospital Clinic, Barcelona and the Graduate School of Medicine, Kyoto University approved this study.
-
Provenance and peer review Not commissioned; externally peer reviewed.