Article Text

Abstract
Objective: To explore the feasibility of prospectively monitoring treatment efficacy and tolerability of infliximab, etanercept, and leflunomide over a two year period in patients with established rheumatoid arthritis (RA) in clinical practice using a structured protocol.
Methods: All patients with RA at seven centres in southern Sweden, for whom at least two disease modifying antirheumatic drugs, including methotrexate, had failed or not been tolerated, who started treatment with either infliximab, etanercept, or leflunomide were included. They were evaluated at predefined times using a standardised protocol including items required for evaluating response to the American College of Rheumatology (ACR) or EULAR criteria. All adverse events were recorded using World Health Organisation terminology. Concomitant treatment and survival while receiving a drug were recorded.
Results: During the study 166 patients were treated with etanercept, 135 with infliximab, and 103 with leflunomide. Treatment response as determined by the ACR and EULAR response criteria was similar for the tumour necrosis factor (TNF) blockers. The TNF blockers performed significantly better than leflunomide both as determined by the response criteria and by survival on drug analysis. Thus 79% and 75% continued to receive etanercept or infliximab compared with 22% of patients who started leflunomide after 20 months. The spectrum of side effects did not differ from those previously reported in the clinical trials. The initial two year experience of a protocol for postmarketing surveillance of etanercept, infliximab, and leflunomide shows that a structured protocol with central data handling can be used in clinical practice for documenting the performance of newly introduced drugs.
Conclusions: Efficacy data for the TNF blockers comply with results in clinical trials, whereas leflunomide appeared to perform worse than in clinical trials. Prolonged monitoring is required to identify possible rare side effects.
- etanercept
- infliximab
- leflunomide
- clinical protocol
- side effects, tolerability
- ACR, American College of Rheumatology
- CRP, C reactive protein
- DAS, disease activity score
- DMARDs, disease modifying antirheumatic drugs
- ESR, erythrocyte sedimentation rate
- HAQ, Health Assessment Questionnaire
- MPA, Medical Products Agency
- RA, rheumatoid arthritis
- TNF, tumour necrosis factor
- VAS, visual analogue scale
Statistics from Altmetric.com
- ACR, American College of Rheumatology
- CRP, C reactive protein
- DAS, disease activity score
- DMARDs, disease modifying antirheumatic drugs
- ESR, erythrocyte sedimentation rate
- HAQ, Health Assessment Questionnaire
- MPA, Medical Products Agency
- RA, rheumatoid arthritis
- TNF, tumour necrosis factor
- VAS, visual analogue scale
Treatment of rheumatoid arthritis (RA) has undergone a dramatic change in recent times with the introduction of new treatments modifying the effects of cytokines. Tumour necrosis factor (TNF) blockade either using a chimeric monoclonal antibody (infliximab) or a recombinant human, soluble TNF receptor (etanercept) is a well documented cytokine modifying principle in clinical use.1–7 Also, other drugs with seemingly well defined modes of action have been introduced. An example is leflunomide, which inhibits de novo pyrimidine synthesis.8–10 For all three treatments the preclinical documentation and experience in clinical trials is extensive.
However, although clinical trials provide important information and are necessary for establishing treatment efficacy and for revealing common side effects, they are not sufficient to establish long term efficacy or to disclose long term or rare adverse reactions.11 Furthermore, the selection criteria normally used for recruiting patients for clinical trials will restrict eligibility to a minority of patients with RA—that is, a lot of patients that we normally see in our clinics will not be included in clinical trials. Therefore, there is a strong case for longitudinal, observational studies using standardised clinical protocols to complement the shortcomings of randomised clinical trials. Such clinical protocols should, despite their open fashion, give information about long term treatment efficacy in a broad spectrum of patients. Importantly, side effects, albeit rare ones or such that occur in particular patient categories, will emerge. In such clinical protocols it is possible to monitor survival while receiving a drug, which reflects both tolerability and efficacy. An advantage of clinical protocols over most clinical trials is that a number of drugs can be monitored using the same protocol, independent of industry support—that is, these drugs can be compared in similar types of patients, avoiding some of the selection bias inherent in the clinical trial. It is time consuming for individual doctors to gain personal experience of new RA treatments, especially if the treatment is given to a small proportion of patients. A standardised clinical protocol shared by a number of subjects, with central data handling and rapid feedback to the treating doctors, would speed up the process of becoming familiar with such new treatments.
We have designed a clinical protocol adapted to monitor new treatments in RA. The objectives of this study were twofold: to investigate the feasibility of the protocol for use in one university centre and in six non-university centres in southern Sweden, and to apply the protocol to evaluate tolerability and efficacy of three new drugs, infliximab, etanercept, and leflunomide, under realistic postmarketing conditions.
PATIENTS AND METHODS
The clinical protocol was developed at the department of rheumatology in Lund and was subsequently approved and used by six other rheumatology units in southern Sweden. The protocol was, in part, built on previous nationwide protocols for early RA monitoring (RAMONA). The quality control character of the protocol made it a part of the legislative documentation required by the authorities in Sweden, and thus no formal approval from the ethical committee was required. The protocol was more comprehensive in Lund in order to allow validation of some of the data from the other centres which were using a more restricted protocol. Thus in Lund a systematic review to ensure that all patients fulfilled the American College of Rheumatology (ACR) 1987 criteria was performed.12
Patient and drug selection
To be eligible for treatment with infliximab, etanercept, or leflunomide, the patients had to fulfil a diagnosis of RA according to the clinical judgment of the treating doctor. All patients included were required to have failed to respond to or not tolerated at least two disease modifying antirheumatic drugs (DMARDs), including methotrexate. The patients were selected on the basis of current disease activity and/or unacceptable steroid requirement as judged by the treating doctor, but had different backgrounds concerning previous treatment, concomitant diseases, and functional impairment and disability. Because one main goal was to include all treatments started, no formal disease activity level other than the doctor's judgment was required and there were no restrictions for systemic or local glucocorticosteroid administration. This is in agreement with the guidelines issued by the Swedish Society of Rheumatology. The patients were included between March 1999 and November 2000 and the study comprises all patients who started treatment with either of these drugs during this period. The last follow up visit included was 1 April 2001.
Because the Swedish legislation allows use of a drug not yet approved by the European Medicines Evaluation Agency (EMEA), we were able, after individual application to the Swedish Medical Products Agency (MPA), to start treatment before the drugs were available in most European countries. The selection of treatment modality depended primarily on drug availability, which varied during the inclusion period. Other factors that influenced selection of drug were variations in resources for intravenous infusions and previous experience of a specific drug during clinical trials in some centres—that is, one centre (Spenshult) specialising in rheumatic rehabilitation had experience of a leflunomide trial and included many patients who were receiving this drug, whereas most other non-university centres have limited inpatient capacity favouring treatment not requiring infusions. Lund recruits patients from primary, secondary, and tertiary care, but patients with RA are mostly recruited from primary care.
The initial doses were as recommended by the manufacturers—that is, 25 mg subcutaneously twice weekly for etanercept, infusion of 3 mg/kg of infliximab at the start of treatment, weeks 2, 6, 12, and thereafter every 8th week. Later the dose of infliximab could be increased and individually tailored when therapeutic response was insufficient, mostly manifested as the patients reporting a postinfusional good response which waned before the next scheduled infusion. The recommendations were first to try increased doses in increments of 100 mg up to a maximum of 500 mg for infusion and keep the same dosage interval. If this failed to give acceptable treatment response the interval was shortened, but more frequent infusions than every four weeks were not allowed. Leflunomide was given as a 100 mg oral loading dose for the first three days and thereafter at a daily dose of 20 mg orally. Patients were allowed to switch between etanercept, infliximab, and leflunomide if withdrawn from any of these treatments. For the assessment of efficacy and tolerability the patient was included in the new treatment group when starting on a new regimen. If restarted on one treatment after a pause the patient was considered to have continued to receive the original therapeutic regimen.
At inclusion the following information was recorded: primary diagnosis, other contributing rheumatological diagnoses, year of onset of primary and other diagnosis, number and name of previous DMARD treatment regardless of treatment length, maximum attained dosage of methotrexate, concomitant DMARD treatment, systemic prednisolone dosage, whether the new treatment was to be given as monotherapy—that is, without any other DMARD, or as an add on (combination) treatment with continuing DMARD treatment. Finally, the treating doctor was asked to make a crude assessment of structural damage and state whether an improvement in the patient's Health Assessment Questionnaire (HAQ) score of 20% or 50% was being aimed at. By aiming for the 20% improvement the doctor indicated that the patient was more severely damaged, and that suppression of the inflammatory component of the patient's disease would have less impact, whereas an expected 50% improvement indicated a smaller destructive component and a dominating inflammatory contribution to the patient's disability. Radiograms were not obtained. Most patients were expected to have advanced disease. Therefore, signs of retarded radiographic progression would be difficult to identify, especially as no formal control group was included.
Assessment
Clinical monitoring included the validated Swedish version of the HAQ,13 28 joint tender and swollen joint count, a 10 cm non-anchored horizontal visual analogue scale for pain (VAS-pain) and general health (VAS-global), the doctor's global assessment of disease activity on a five grade scale (Dr-global), erythrocyte sedimentation rate (ESR) according to Westergren, and C reactive protein (CRP).14 This set of variables was obtained at 0, 3, 6, 12 months (optional 0.5, 1.5, and 9 months), and thereafter every 3–6 months. No patients were excluded if registrations were missing, except for the mandatory inclusion variables at time point 0. All registrations of patients treated with infliximab were made immediately before infusion. The study is continuing, hence patients included have, at the defined time points in this report, been followed up for different lengths of time. Withdrawal from treatment was classified as withdrawal caused by adverse reaction, lack of response, or other. All centres were urged to monitor carefully and report development of side effects using forms from the Swedish MPA conforming to the World Health Organisation adverse reaction terminology.
A computer application was developed (PC-milieu, MS Access)15 to calculate the disease activity score for the 28 joint indices (DAS28).14 The program also calculated whether the patient improved as defined by the EULAR criteria based on DAS28 by classifying the patient into one of the three groups non-, moderate, or good responder, and also if the remission criteria (DAS28<2.6) were met.14 Classification according to the ACR20, 50, and 70 response criteria was also calculated by the application.16 These results together with a graphical presentation of the clinical and laboratory variables over time were promptly forwarded to the treating doctor from the centralised computer handling. The application also compiled the overall results, as well as results for individual centres. The application presented survival while receiving a drug graphically. To improve compliance the application also produces reports of missing follow up visits, which is forwarded to the treating doctor.
Statistical calculations
Differences between groups were analysed by the χ2 test for ordinal variables and the Mann-Whitney U test for numerical variables. The Wilcoxon matched pair sign rank test was used for comparison within groups. A p value of <0.05 was considered significant. Comparison of patients continuing the different treatment regimens (survival on a drug) was calculated at defined times using χ2.
RESULTS
During the study 369 patients were treated. Of these, 33 patients tried two treatment modalities, and one tried all three drugs. Thus 404 treatments with any of these drugs were started. Etanercept was given to 166 patients, infliximab to 135 patients, and 103 were treated with leflunomide.
Table 1 summarises the demographic data for patients receiving each treatment regimen. Note that some patients may appear in more than one group. Table 2 compares some characteristics of the patients treated in Lund with those of the patients from the other centres. Of the 142 patients treated in Lund, 90% were rheumatoid factor positive, 88% were erosive, 38% had nodules, and 98% fulfilled the ACR RA classification criteria.
Patient characteristics (n=369) according to therapeutic regimens (n=404). The numbers and values relate to the treatments—that is, a patient may be included more than once
Patient characteristics at the start of treatment in Lund compared with other centres. The numbers and values relate to the treatments—that is, a patient may be included more than once
Patients treated with the TNF blockers shared the same characteristics, differing only in the number of continuing DMARDs and thus in monotherapy or combination therapy. In contrast, patients treated with leflunomide were older, seemed to have more severe joint damage, were more often treated with monotherapy, and had somewhat lower inflammatory activity. Similar differences were, as expected, apparent in the comparison between Lund and the other centres because only 13 patients were treated with leflunomide in Lund.
Figures 1A–C show the treatment efficacy as measured by the ACR20, 50, 70 response. The responses according to the EULAR criteria using DAS28 were similar (data not shown). Etanercept was significantly better than infliximab at three months (p<0.02) and six months (p<0.05) when the number of patients reaching the ACR20 response were compared. For the ACR50 response only the three months' registration reached significance in favour of etanercept compared with infliximab (p<0.05). No difference was found between these regimens at 0.5, 1.5, 9, and 12 months using the ACR20 and the ACR50 response criteria. When patients reaching both the ACR20 and ACR50 response were compared, leflunomide was significantly less effective than etanercept at three months (p<0.001) and six months (p<0.05). Infliximab was better than leflunomide only at three months for ACR20 (p<0.01) and ACR50 (p<0.05). The limited number of observations for leflunomide at the other time points made statistical comparisons unreliable.

Percentage of patients fulfilling the ACR20, ACR50, and ACR70 response at follow up times for (A) etanercept, (B) infliximab, and (C) leflunomide. For each follow up time and treatment regimen all the ACR70 are included in the ACR50, which in turn are all included in the ACR20 response patients. For statistical comparison between the treatment regimens see “Results”.
When the ACR20 and the ACR50 criteria (data not shown) were used, the treatment response for patients from Lund treated with etanercept or infliximab did not differ from that of patients from the other centres receiving these treatments. The limited number of patients treated with leflunomide in Lund made comparison with the other centres unreliable.
Figures 2A–C depict the change in prednisolone dosage during treatment. For both etanercept and infliximab a significant reduction could be made already after two weeks (p<0.001) and the prednisolone dosage continued to decrease during the first 12 months for etanercept. For infliximab the maximal reduction was reached after three months of treatment. In contrast, no reduction of prednisolone dosage was possible in the leflunomide group except in the small subset of patients continuing to receive the treatment at 12 months (p<0.05 at 12 months).

Tenth, 25th, median, 75th, and 90th centiles of the prescribed weekly prednisolone dosage at follow up times for (A) etanercept, (B) infliximab, and (C) leflunomide. *p<0.05, **p<0.01, ***p<0.001, NS, not significant compared with baseline values at time 0.
Patients defined as more or less damaged at the outset of the study (expected improvement in HAQ 20% or 50%) showed no difference in response for either etanercept, infliximab, or leflunomide when tested after three and six months of treatment using ACR or EULAR response criteria (data not shown).
Response rates of infliximab and etanercept used as monotherapy were not statistically different from the combination therapy with one or more DMARDs after three and six months of treatment (data not shown).
Forty three per cent of the patients receiving infliximab responded to the original dosage regimen. However, in 57% of the patients an increased dosage or shortened interval, or both, was required for symptomatic control.
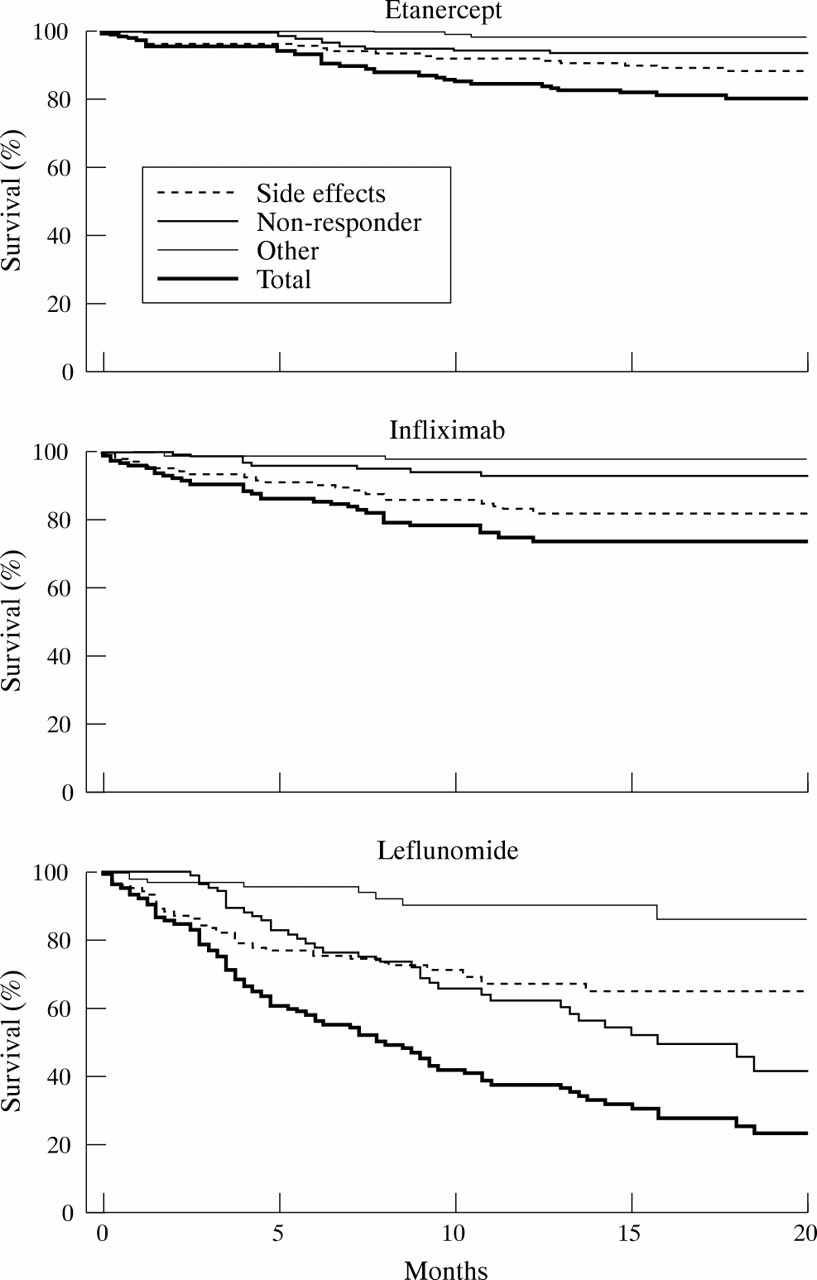
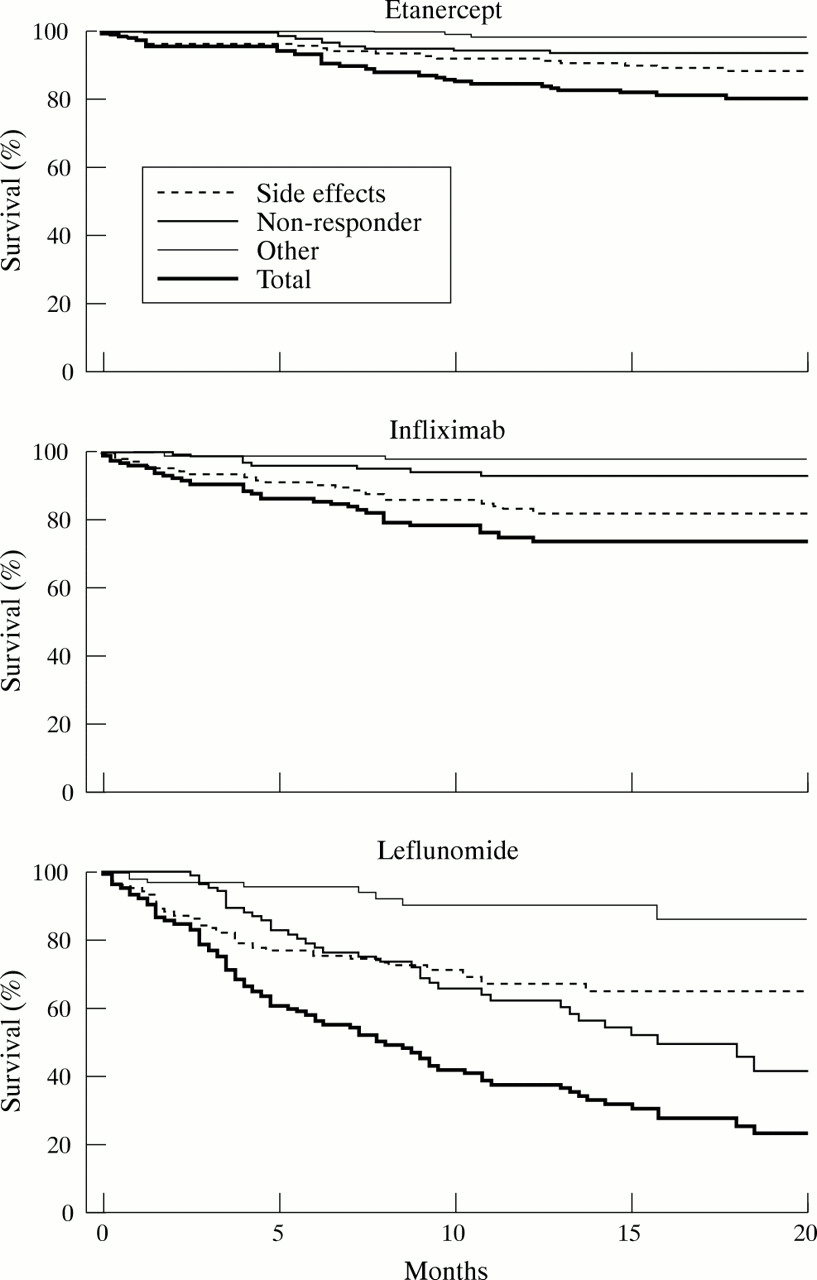
Figures 3A–C shows the “survival while receiving a drug”—that is, the continuation of the respective treatment. There were no significant differences between etanercept and infliximab, with 79% and 75% of patients, respectively, continuing to receive the drug after 20 months. In contrast, only 22% of patients were still receiving leflunomide after 20 months (p<0.001 versus both etanercept and infliximab). Withdrawals were significantly more common for leflunomide as early as five weeks (p<0.05 versus etanercept and infliximab). During the initial six months leflunomide withdrawal was mostly due to side effects. Lack of response was the major reason for stopping the drug after longer exposure. For infliximab and etanercept adverse reactions were the main cause of drug withdrawal throughout the study.
{kind=link}
{kind=link}
{kind=link}
Survival while continuing to receive etanercept, infliximab, or leflunomide. For statistical comparison between the treatment regimens see “Results”.
Table 3 summarises the total numbers of adverse reactions reported, according to severity and in relation to the time of observation for the different treatments. The total numbers of observational years were 232.8, 111.1, and 70.9 for etanercept, infliximab, and leflunomide respectively.
Graded side effects per 100 treatment-years. The grading was done by the treating doctor on the Swedish Medical Products Agency forms
The fatal adverse reactions in the patients receiving etanercept were one gastroenteritis—day 180, one immunocytoma of the breast—day 220, one myocardial infarction—day 413. The serious adverse reactions in the etanercept patients were four myocardial infarctions—days 41, 63, 130, 501, three bacterial infections (two pneumonia including one with septicaemia, one septic arthritis)—days 130, 150, 270, two uterine cervical carcinoma (one in situ)—days 160, 413, one acute myeloic leukaemia—day 440, one general malaise—day 350, one leucopenia—day 91, one Bell's paralysis—day 130, one cutaneous vasculitis—day 368, one discoid lupus (recurred on provocation, also provoked by infliximab)—day 69.
The life threatening adverse reactions in the infliximab patients were one anaphylactoid reaction—day 320, one mesothelioma—day 42, one pharyngitis with extremely severe spread to the throat, neck, and upper abdomen with compression of the upper airways—day 480. The serious adverse reactions in the infliximab patients were four allergic reactions—days 41, 201, 230, 573, two bacterial infections (one otitis media, one cystitis)—days 108, 210, one Hodgkin lymphoma—day 129, one non-Hodgkin lymphoma—day 180, one thrombocytopenia—day 250, one lupus like reaction—day 230, one discoid lupus—day 20. The five serious allergic reactions seen during infliximab treatment were all in patients given combination therapy including methotrexate. Of the two autoimmune reactions seen, the discoid lupus was sequentially provoked by both etanercept and infliximab. The other reaction consisted of fever, rash, leucopenia, positive antinuclear antibody and anti-DNA tests, and resolved completely clinically and as measured by the laboratory variables when infliximab was withdrawn.
The serious adverse reactions in the leflunomide patients were one leucopenia—day 108, one deep vein thrombosis with hypertension and visual impairment—day 226, one throat pain with swelling of tongue and difficulties in swallowing and loss of taste—day 60, one clinical polyneuropathy with paraesthesias in feet and shoulders—day 110.
DISCUSSION
We here report the first results of a clinical protocol for centralised monitoring of the performance in clinical practice of three recently introduced drugs. Some key observations pertaining both to the approach itself and to the results obtained need special attention.
Firstly, we wanted to see whether the protocol could be used both at a university centre and at six other rheumatology units with less developed resources for data management. The protocol, constructed according to the design of the protocol widely used for early RA monitoring in Sweden, was well accepted by the participating centres. By the more comprehensive evaluation of the Lund patients and by comparing the Lund cohort with the cohort from the other units, we showed that the protocol could be applied at both types of centre. The data obtained in Lund conformed overall with the data obtained at the other centres, although the characterisation of the patients not at Lund was less detailed. The restricted amount of information gathered contributed to the good quality of input data, and the regional basis ensured that a network for supplementary information was easily achieved. Centralised data entry ensures uniform interpretation of information on forms. The more simple protocol thus seems suitable for future use and could be implemented in clinical practice.
One main finding of the study was that the performance of the two TNF blockers complied with results in published clinical trials, albeit with a somewhat lower response rate.1–7 Leflunomide, on the other hand, performed less favourably as measured by efficacy variables and survival on the drug.8–10,17 This may be due, in part, to patient selection, as most of the leflunomide patients were recruited from one centre. Thus, direct comparisons should be interpreted with caution. However, both the efficacy variables, which according to definition only assess those patients continuing to receive the drug—that is, those who respond most favourably, and the survival on drug data significantly favour the TNF blockers. This suggests that although the groups have somewhat different characteristics (table 1), the drugs performed differently in these patients with longstanding RA.
The open character of a clinical protocol may possibly induce a placebo effect and bias favouring a positive response, but this is not likely in view of the results, which are not better than the results of clinical trials. Furthermore, a possible bias is probably similar for all treatments, making comparisons more relevant. A confounding factor for interpretation of the results is the unrestricted use of glucocorticosteroids. This introduces the disadvantage of enrolling patients, most of whom were treated with glucocorticosteroids before enrolment, and who in a considerable number of cases had had increased doses while waiting for the new treatment to start. This would underestimate the treatment efficacy of the new drug. On the other hand, continuing glucocorticosteroid treatment made it possible to evaluate treatment response also by monitoring tapering of this treatment (figs 2A–C).
Minor differences in performance were seen between the TNF blockers. A somewhat better response rate for the ACR20 was found for etanercept at the three and six month follow up only. Adjustments of the dose were necessary for 57% of the infliximab patients. Thus, the waning efficacy of infliximab during these treatment intervals may influence the comparisons. At most times, no difference between the two TNF blockers was found.
An important goal of a clinical protocol is to facilitate recording of side effects and identification of adverse reactions not previously seen in clinical trials. We found a spectrum of side effects, but these did not differ from those previously reported.1,10,18 However, it should be noted that three lymphomas and one case of acute myeloic leukaemia were seen in this limited patient cohort treated with TNF blockers.
As in clinical trials, we recorded all events regardless of whether a causal relationship with the drug was suspected. Continued long term monitoring of the included patients and recruitment of new patients into the protocol is necessary to enable full evaluation of the spectrum of side effects. To identify rare side effects, including specific types of malignancies, it is necessary to combine data from this continuing protocol with data from other protocols nation wide.
REFERENCES
Footnotes
-
The South Swedish Arthritis Treatment Group includes Helsingborg: Kristina Forslind, Gunilla Gunnarsdotter, Göran Karlsson, Catarina Keller, Sven Noltorp, Dick Sahlberg. Kristianstad: Ido Leden, Jan Theander. Lund: Anita Åkesson, Izabella Bartosik, Christine Bengtsson, Ulf Bergström, Meliha Crnkic, Rabi Dash, Eva Edlund, Renate Elborgh, Pierre Geborek, Andreas Jönssen, Åsa Hägglund, Lotta Larsson, Elisabeth Lindqvist, Bengt Månsson, Ola Nived, Gabriella Olsson, Lars Rydgren, Tore Saxne, Agneta Scheja, Christina Ståhl-Hallengren, Gunnar Sturfelt, Gertrud Widell. Simrishamn: Anders Gülfe, Helén Petri. Spenshult: Torvald Berg, Ann Helén von Braun, Julio Goobar, Gunilla Holmström, Leena Niemelää, Olle Nillius, Ingemar Petersson, Per-Erik Rönnborg, Hanna Slomkowska, Annika Teleman. Trelleborg: Miriam Karlsson, Eva Nitelius. Växjö: Anneli Östensson, Maria Söderlin.