Article Text

Abstract
OBJECTIVES Reversible posterior leucoencephalopathy syndrome (RPLS) may develop in patients with renal insufficiency, hypertension, and immunosuppression, and is managed by prompt antihypertensive and anticonvulsant treatment. Four patients with renal insufficiency and fluid overload associated with Wegener's granulomatosis (one patient) and systemic lupus erythematosus (SLE) (three patients) are described, whose clinical picture and neuroimaging indicated RPLS.
CASE REPORTS All patients had headache, seizures, visual abnormalities, and transient motor deficit, and were hypertensive at the onset of the symptoms. Head computed tomography (CT) scan and magnetic resonance imaging showed predominantly posterior signal abnormalities, which were more conspicuous on T2 weighted spin echo images than on CT scan. All patients had some form of cytotoxic treatment shortly before the syndrome developed, and dramatically responded to blood pressure control and anticonvulsant treatment. In two patients with SLE, dialysis was required for renal insufficiency.
DISCUSSION Follow up neuroimaging studies showed almost complete resolution of signal abnormalities, and suggested that RPLS was associated with cerebral oedema without concomitant infarction. The treatment of hypertension and neurotoxic condition such as uraemia appears of primary importance, while immunosuppressive treatment may cause further damage of the blood-brain barrier.
- reversible posterior leucoencephalopathy syndrome
- systemic vasculitis
- connective tissue disease
- systemic lupus erythematosus
Statistics from Altmetric.com
- reversible posterior leucoencephalopathy syndrome
- systemic vasculitis
- connective tissue disease
- systemic lupus erythematosus
Reversible posterior leucoencephalopathy syndrome (RPLS) is a clinicoradiological syndrome, first described by Hincheyet al in 1996,1 that can be associated with several conditions, including hypertensive encephalopathy, chronic renal insufficiency, blood transfusion, puerperal eclampsia.1 ,2 RPLS may also develop during treatment with cisplatin,3 immunosuppressive drugs such as cyclosporin A,4 tacrolimus, interferon α,5immunoglobulin,6 and after liver transplantation7 or the acute phases of autoimmune diseases.1 Hypertension has often been emphasised as a common feature of all RPLS associated conditions.1 ,5 ,8-11
The most common clinical manifestations of RPLS are headache, nausea and vomiting, altered mental status, decreased alertness, seizures, cortical blindness and other visual abnormalities, and transient motor deficits.1 The main finding in neuroimaging is posterior white matter oedema, often with a strikingly symmetrical involvement of the parietal and occipital lobes.1
After reviewing three years of neurological consulting at the Department of Internal Medicine of Genoa University we identified four patients (which corresponds to about 0.1% of all observed cases) with systemic lupus erythematosus (SLE) or vasculitis whose clinical and neuroimaging features indicated a diagnosis of RPLS .
Case reports
CASE 1
A 23 year old man presented to the emergency room with a first tonic-clonic seizure. He had a history of Wegener's granulomatosis since the age of 20, presenting with respiratory and renal symptoms. The diagnosis was based on American College of Rheumatology (ACR) criteria including nasopharyngeal biopsy. A previous episode of renal failure in the course of the disease had been treated with prednisone 1 g/day for two days and cyclophosphamide 750 mg/day for five days, with clinical improvement. Renal biopsy indicated diffuse extracapillary glomerulonephritis. Six days after the first seizure, he developed headache, vomiting, nausea, diplopia, and recurrent seizures, followed by stupor and right hemiparesis. Blood pressure increased to 220/150 mm Hg, and serum creatinine was 320 μmol/l. A CT scan of the brain showed diffuse occipital hypodensity. MRI showed T2hyperintense and T1 hypointense signal abnormalities in the occipital lobes bilaterally. He received intravenous antihypertensive and anticonvulsant treatment. Three days later his neurological findings returned to normal. The EEG showed moderate bilateral slowing in the occipital regions. Repeat MRI and EEG two weeks later were unremarkable.
CASE 2
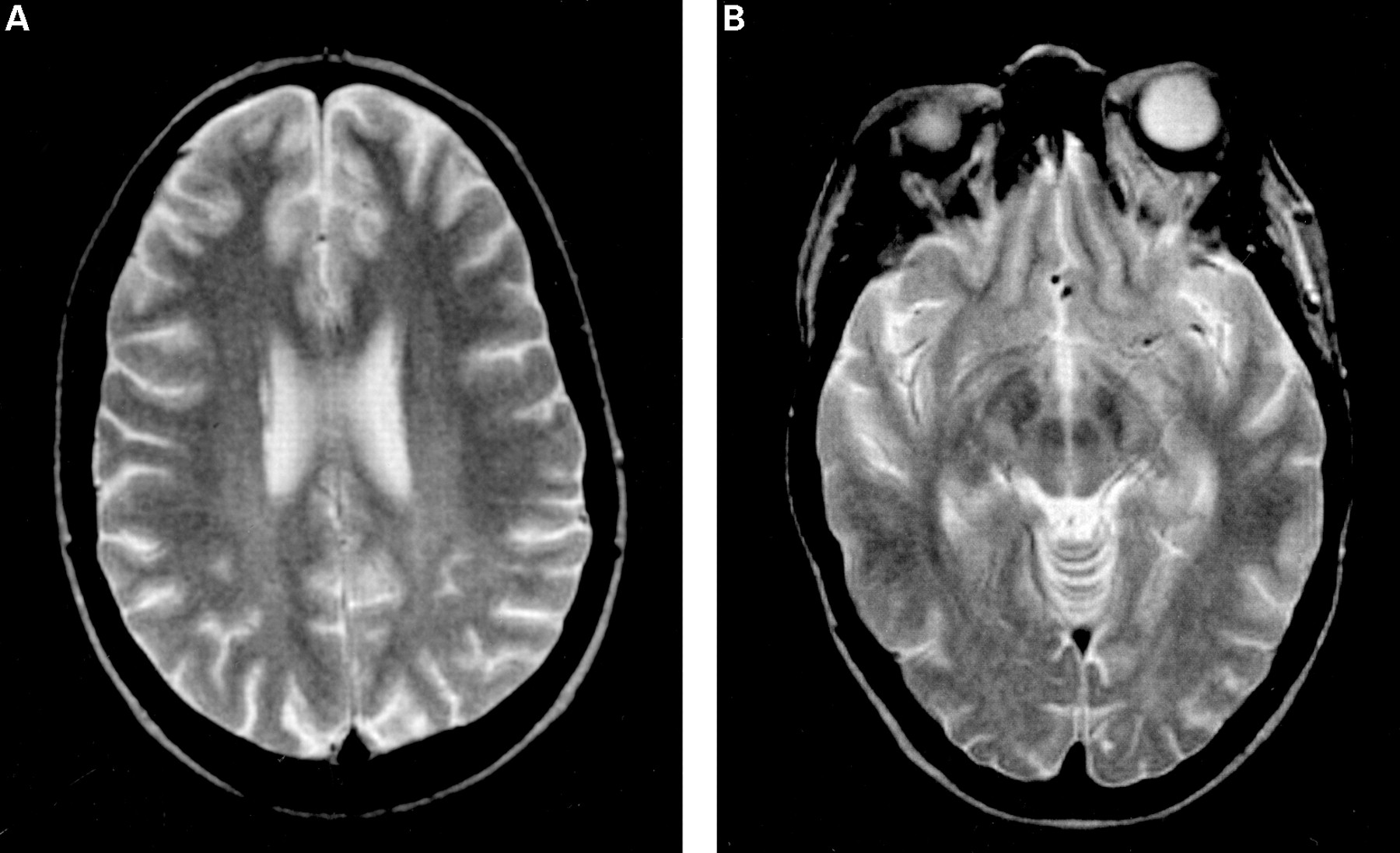
A 22 year old woman was admitted to the Internal Medicine Department owing to renal insufficiency. She had a history of nephritis secondary to SLE, renal failure, and hypertension since the age of 17. The subsequent rapid deterioration of renal function with severe hypertension was treated with intravenous cyclophosphamide and methylprednisolone, with some clinical improvement. Renal biopsy indicated lupus membranous glomerulonephritis with diffuse proliferative lesions. One week after the admission she presented sudden frontal headache, visual blurring, and altered mental functioning. Blood pressure was 200/130 mm Hg and serum creatinine was 390 μmol/l. Twenty minutes after the onset of headache she had a partial seizure with motor symptoms, and was given intravenous antihypertensive treatment and clonazepam. Transient episodes of visual blurring recurred several times during the same day. Six hours after the onset, she was still reporting headache; her neurological status indicated mild left hemiparesis, hyperreflexia, and diplopia to the left. The EEG showed severe generalised discharges more evident over the occipital regions. MR cranial images disclosed T2hyperintensity in bilateral frontal, occipital, and posterior parietal lobes (figs 1A and B), right posterior temporal lobe, and cerebellum. Four days later the neurological examination was normal. There were no seizure recurrences, and follow up brain MRI (figs 2A and B) and EEG showed complete resolution.

Brain MR scan of patient 2 in the acute stage of the disease: T2 weighted images show widespread white matter signal abnormalities, which particularly involve frontal, occipital (A) and parietal (B) lobes.

{kind=link}
{kind=link}
Follow up brain MR scan of patient 2, showing the disappearance of the white matter signal abnormalities.
CASE 3
A 22 year old woman was admitted to the hospital for fever, headache, and polyarthralgia. She had been diagnosed as having SLE one year before, and treated since then with corticosteroids and azathioprine. On the second day of admission to hospital she developed acute renal failure with serum creatinine level increasing from baseline values of 340 μmol/l to 840 μmol/l. Blood pressure was 170/110 mm Hg. Lupus nephritis was diagnosed on clinical grounds, but renal biopsy was not performed in view of the critical condition. She received bolus doses of methylprednisolone for three days followed by cyclophosphamide 500 mg for two days. During the treatment she presented severe headache and recurrent seizures, followed by visual blurring and altered mental functioning. The EEG showed severe generalised dysrhythmia. MRI showed symmetric T2hyperintensity in the white matter of occipital, posterior parietal, and temporal lobes. She was given antihypertensive and anticonvulsant treatment, and underwent haemodialysis until renal dysfunction improved. A neurological examination 10 days later was unremarkable and a repeat MRI was normal.
CASE 4
A 30 year old woman presented with fever, polyarthralgia, and diffuse weakness. She had a history of SLE with renal and haemopoietic dysfunction since the age of 20; renal biopsy indicated lupus nephritis. She had received cyclophosphamide for six years and then prednisone, with some clinical improvement. After admission to the Internal Medicine Department, treatment with intravenous corticosteroids was started. Eight days later she presented headache, drowsiness and a tonic-clonic seizure, while her blood pressure increased to 210/125 mm Hg. She was given intravenous diazepam; after the seizure, her neurological status indicated stupor, slowed responses, diffuse hyperreflexia, and a right Babinski sign. At the same time, serum creatinine rapidly increased to 550 μmol/l and blood urea nitrogen to 37.5 mmol/l of urea. She was given sodium nitroprusside and haemodialytic treatment was started. On the next day, neurological findings still indicated drowsiness, altered mental functioning and visual blurring. Brain CT scan showed diffuse hypodensity in bilateral occipital and frontal regions. Two weeks later blood pressure and serum creatinine returned to baseline values, visual blurring cleared and the neurological status was unremarkable. A follow up CT scan was normal.
Discussion
Table 1 summarises the clinical and neuroimaging findings of the four reported patients. All patients developed a reversible syndrome consisting of headache, seizures, visual abnormalities, and transient motor deficit, whose onset was marked by severe hypertension. MRI of the brain showed predominantly posterior signal changes; in the patient who underwent both CT scan and MRI, the abnormalities were more conspicuous on T2 weighted spin echo images than on CT scan. Therefore all patients met the clinical and radiological criteria of RPLS diagnosis. Follow up neuroimaging studies showed almost complete resolution of signal abnormalities, confirming that RPLS was associated with cerebral oedema without concomitant infarction.
Summary of clinical and neuroimaging findings of the reported patients
RPLS is a clinicoradiological entity and it is only since the introduction of modern scanning methods that the posterior parieto-occipital lesions have been linked with the clinical features described.9 Although the frequent association of RPLS with severe hypertension may suggest some conceptual overlapping between this syndrome and hypertensive encephalopathy, a confusion between the two clinical entities is to be avoided; in fact RPLS may occur in several conditions, even in the absence of severe hypertension.5
The hypothesis that best explains the acute and reversible white matter changes in RPLS is that hypertension induces a loss of cerebral autoregulation with secondary vasodilatation, increase of vascular permeability, petechial haemorrhages, and extravasation of fluid into the brain, especially in arterial boundary zones.1 ,5 The parieto-occipital regions are predominantly affected, which may depend on the differences in autonomic innervation as compared with anterior circulation.5
Fluid retention secondary to renal failure, or the cytotoxic effects of immunosuppressive agents, may cause a dysfunction of vascular endothelium, thus impairing autoregulation and causing encephalopathy, whenever even a slight increase of blood pressure concurs.5
It is noteworthy that all patients had some form of cytotoxic treatment (often high dose) shortly before the syndrome developed. On the other hand, they also had signs of acute renal insufficiency developing against a background of a chronic pathology related to the connective tissue disease. Although it is difficult to ascertain the causal role of immunosuppressive drugs as compared with hypertension and fluid overload resulting from renal failure, it is conceivable that both mechanisms contributed to the damage of the blood-brain barrier, with a consequent increase of oedema.1
Only four cases of RPLS associated with connective tissue disease have been reported so far, at least to our knowledge. Two of them had a history of SLE,1 and two had a diagnosis of polyarteritis nodosa.12 ,13 Also RPLS in patients with autoimmune disease is likely to depend on a dysfunction of vascular endothelium secondary to multiple factors such as hypertension, renal failure with fluid retention, and cytotoxic drugs. This is confirmed by the dramatic effect of prompt antihypertensive treatment, associated with removal of fluid overload by haemodialysis in two patients.
Some authors emphasise that seizures may cause or aggravate vasogenic oedema in a brain in which autoregulation has been disrupted, and that seizures may herald the onset or represent the major clinical manifestation of RPLS.14 ,15 Seizures occurred before RPLS in all patients, and recurred during the clinical course in three of them.
The clinical findings and the CT imaging of RPLS are not quite specific, so that a differential diagnosis with several neurological conditions may be difficult, including simultaneous bilateral infarction of the posterior cerebral artery, cerebral venous sinus thrombosis, demyelinating disorders or encephalitis, and cerebral vasculitis. Neuroimaging in cerebral vasculitis, however, may be unremarkable or show atrophy and focal ischaemic infarcts affecting several vascular territories, rather than white matter abnormalities in the posterior cerebral regions. Anyway, the most appropriate investigation in RPLS is MRI, and particularly diffusion weighted MRI, which may help to discriminate the vasogenic oedema of RPLS and the cytotoxic oedema of early cerebral infarction.5
RPLS must be recognised early because of potential reversibility with prompt antihypertensive treatment. The management is therefore different from that of ischaemic stroke, where recent guidelines warn against treating mild and moderate hypertension.5 These patients are usually admitted to hospital in internal medicine departments, and internists should be aware of this when managing patients with autoimmune disease and sudden onset of encephalopathy associated with posterior white matter changes on neuroimaging.
In conclusion, we feel that RPLS associated with connective tissue disease results from a concurrence of multiple factors, and requires prompt treatment of both hypertension and seizure, associated with haemodialysis in the case of acute renal failure. On the contrary, immunosuppressive agents, which are normally used in the exacerbations of the underlying disease, are potentially dangerous, and we suggest that the use of these drugs is deferred until the acute stage of the syndrome is over.