Article Text

Abstract
Rheumatoid arthritis (RA) is characterised by both inflammation, as manifested by pain and swelling, and destruction of the joints. Unequivocal evidence indicates that disease activity, and thus the inflammatory response, is linked to joint damage. From this viewpoint we suggest that, vice versa, joint damage might be a cause of the active disease process, thus leading to a vicious cycle of events. The background to this notion stems from the known autoimmune response in RA, the potential of cartilage and bone breakdown products to elicit inflammation and notions that in joints that have undergone surgery with cartilage removal RA does not flare. However, the clinical evidence for this relationship is still to be provided as proof of the concept.
Statistics from Altmetric.com
Rheumatoid arthritis (RA) is characterised by inflammation of the synovial membrane, which goes along with destruction of bone and cartilage. The degree of the inflammatory disease activity is highly related to the grade of physical functional impairment,1 but the accrual of joint damage over time is also contributing importantly to disability.1 2
Current evidence suggests that the extent of inflammation governs the development and level of joint damage. In this regard, disease activity according to composite disease activity indices is a good predictor of the degree of joint damage.3 This relationship also pertains to swollen joint counts4 and acute phase reactant (APR) levels.5 In addition, however, the presence and titre of rheumatoid factor (RF) and the presence of antibodies to citrullinated proteins (ACPA)6–9 are related to the evolution of joint destruction. Moreover, the extent of joint damage at the start of an observation period is also predictive of its subsequent progression.9 10 How do these various findings fit to each other and into a pathogenic concept?
THE TRADITIONAL PARADIGM: INFLAMMATION LEADS TO DESTRUCTION
The cause of RA is unknown and many different events may be involved in triggering the pathogenic process.11 12 These events may comprise an initial activation of the innate immune system and presentation of disease eliciting antigens by antigen presenting cells (APC) to T lymphoyctes. This process apparently does not undergo the usual self-limitation characteristic of normal immunoinflammatory reactions, rather chronicity ensues. In addition, B cells, themselves also subserving antigen-presenting function, produce autoantibodies that may contribute to the process by formation of immune complexes that in turn can activate complement and inflammatory cells. Whether these early steps are closely interrelated and interdependent or rather constitute distinct events that are not or only minimally linked, reflecting the heterogeneity of RA, is yet unknown. However, the final pathways appear to be common in most patients, namely a vigorous inflammatory process characterised by the production of the proinflammatory cytokines tumour necrosis factor (TNF), interleukin (IL)1 and IL6; at the end of the pathogenic cascade stand synovial inflammation by virtue of infiltration of numerous cells and cell populations and mediator release, cartilage degradation induced by matrix metalloproteinases, and bone erosions mediated by osteoclast activation, all these phenomena being orchestrated by the proinflammatory cytokines.11–13
Over the past two decades, clinical and basic rheumatology research have focussed on the inflammatory component of RA and, on the basis of extensive evidence partly mentioned above, defined this component as the culprit of the most important outcomes of RA, joint destruction and disability. This has led to the description of a direct relationship between inflammation and joint destruction and disability on the one hand, and of joint damage with disability on the other hand (fig 1).1 2 14 However, in other diseases, such as reactive or psoriatic arthritis, synovial inflammation is histologically similar to that of RA without eliciting a similar degree of joint destruction as in RA.15 Moreover, acute phase reactant levels are also akin among these diseases.16 Thus, the evolution of the destructive changes typical of RA must involve additional pathways beyond mere inflammation. These pathways can be hardly explained by the qualitative involvement of inflammatory cytokines, since the same cytokines are upregulated in all these diseases,17 as also shown by the response to therapies targeting cytokines.

RHEUMATOID ARTHRITIS AS AN AUTOIMMUNE AND IMMUNE COMPLEX DISEASE AND THE ROLE OF TISSUE DAMAGE
One characteristic of RA differentiating it from other inflammatory joint diseases is the presence of autoantibodies, particularly RF and ACPA. These have been more recently brought into the context of being associated with disease activity, although their presence has been found related to the degree of joint damage.7 18 However, the mere existence of the autoimmune response may not be sufficient to drive the inflammatory disease process, since autoantibodies can be detected up to 10 years before the development of the first symptoms of RA.19 Therefore, in order to encapsulate the various associations into a more unifying concept, other aspects may require consideration.
It has long been known that immune complexes are deposited in RA cartilage,20 that complement is activated in RA synovial fluids as a reflection of the presence of immune complexes,21 that Fcγ deficiency ablates experimental RA-like arthritis,22 and that immune complexes activate proinflamatory cytokines.23 The immune complexes of patients with RA may contain RF, ACPA, collagen and anti-RA33 and can be present in the circulation and in joint fluids.24–27 Moreover, the presence of autoantibodies has been found to be associated with the major genetic characteristic of RA, the shared epitope,28 and citrullination of proteins appears to increase the potential for antigen presentation.29 The subsequently augmented cytokine production and inflammatory response may constitute the link to the association of autoantibodies with increased joint damage.
Several decades ago, rheumatologists have observed that, beyond the production of RF, patients with RA exhibit an immune response to connective tissue breakdown products. For example, collagen has been regarded to be a major autoantigen driving the immunoinflammatory response in RA.30 These notions have been supported by observations of humoral and cellular immune responses to collagen,31 32 the presence of anti-collagen antibodies trapped in articular cartilage from patients with RA,20 and the finding that collagen can elicit a severe destructive form of experimental arthritis.33 This arthritis can also be mediated by anti-collagen antibodies. Indeed, in its course, presumably as a consequence of immune complex formation and posttranslational antigen modifications, other autoantibodies such as RF and ACPA may occur.30 34 Moreover, free collagen itself can activate a macrophage response.35 Aside from collagen, other cartilage constituents can induce experimental arthritis.36 Thus, Koch’s postulate as adopted for autoimmune diseases37 is at least partly fulfilled in this context.
Proteases responsible for much of the damage observed in arthritic cartilage and bone, release breakdown products and can liberate a variety of neoepitopes.38–40 Such neoepitopes are often used as biomarkers thought to reflect tissue damage. Neoepitopes may also ensue by posttranslational modifications including citrullination.41
In further support of the contribution of autoimmunity to the disease process, in a solely TNF-triggered experimental arthritis model, autoimmunity to an autoantigen associated with RA, the heterogeneous nuclear ribonucleoprotein A2 (the RA33 autoantigen) was observed, the molecule was aberrantly expressed in the joint and early immunisation of the TNF transgenic (TNFtg) mice with RA33 augmented the arthritic process.42
Taken together, these data are compatible with the assumption that a trigger that leads to tissue damage and liberation of neoepitopes in connective tissue proteins may elicit an (auto)immune response and lead to the subsequent activation, perpetuation and/or chronification of arthritis. Thus, the mere induction of cartilage or bone damage may be followed by a cascade of autoimmune and inflammatory changes that induce a further increase in inflammatory cytokines, leading to quantitative differences when compared with other arthritides.17
THE REVERSAL OF THE CLINICAL PARADIGM: DOES DAMAGE CAUSE DISEASE ACTIVITY?
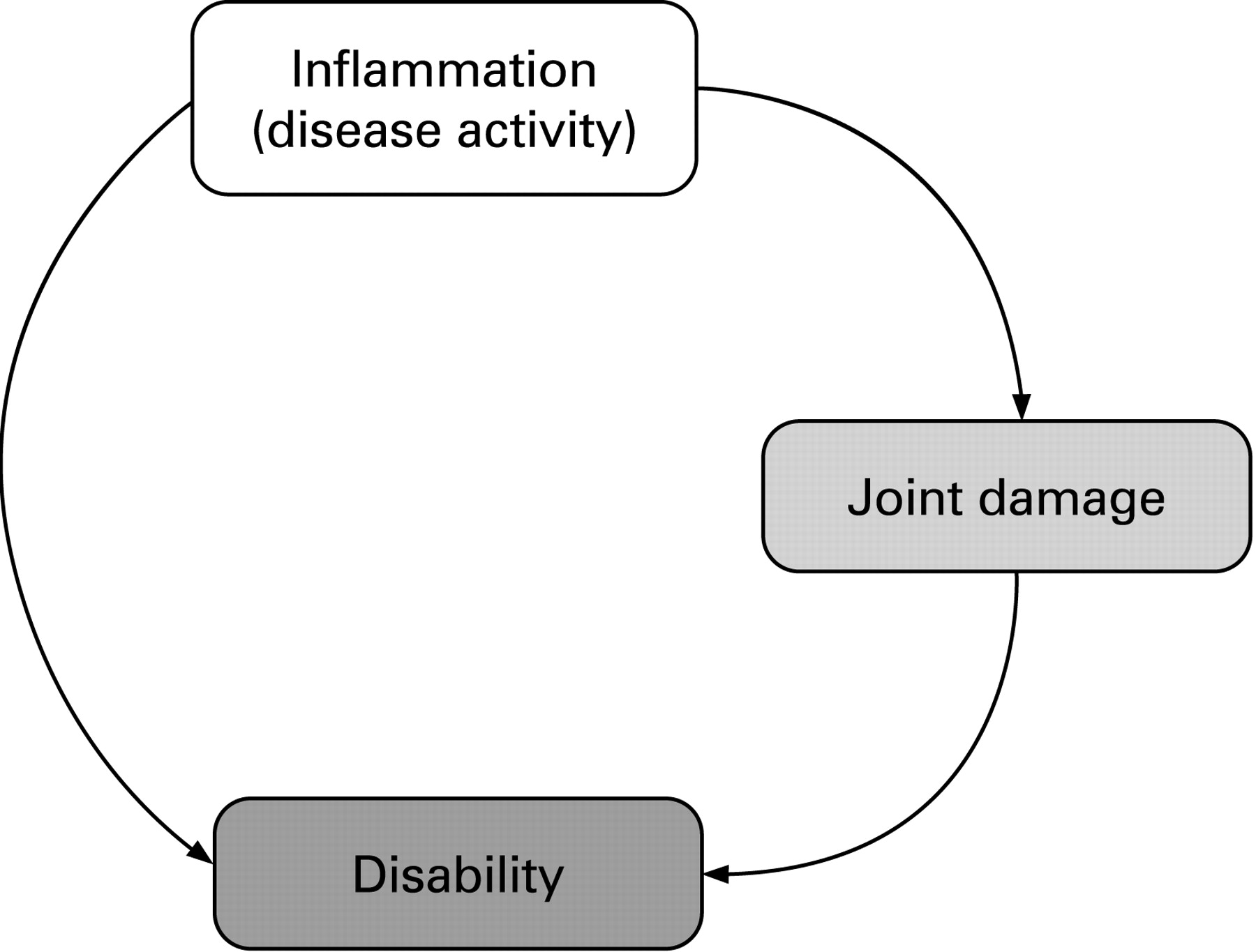
On the basis of the evidence discussed above and suggestions that cartilage autoantigens may drive chronic synovitis,43 it has been reasoned44 and can be hypothesised that joint damage activates inflammatory processes (fig 2). This incident would constitute the true vicious cycle of RA.

{kind=link}
{kind=link}
In line with this hypothesis are observations that surgical or radiation synovectomy is frequently associated with relapses of RA, at least once cartilage damage is present.45–48 By contrast, elimination of articular cartilage with total joint replacement or cartilage shaving decreases the frequency of relapses and leads to significant reduction in synovitis.47 48 The fact that 86% of psoriatic arthritis knees, but only 46% of RA knees were in remission at 36 months after arthroscopic synovectomy,48 supports the notion that in RA, in contrast to PsA, other elements than events just confined to the synovial membrane may drive the disease. Indeed, Konttinen et al had observed that commonly “synovitis reoccurs after chemical or surgical synovectomy after an initially successful operation”.47 They contrast this to their clinical observation “that chronic synovitis rarely reoccurs in joints from which articular cartilage has been totally removed” and that they saw synovitis only when “the patellofemoral joint has not been replaced with a patellar implant, and therefore part of the hyaline articular cartilage has remained in situ”.47 In support of these clinical observations, they show that the synovial membrane of joints undergoing revision arthroplasty after full cartilage removal during the first operation lacks activated T cells, in contrast to the primary synovial membrane.47 Although the clinical observations they described were not the major topic of this study, their data provide indirect evidence for their assumption. To bolster their annotation, it would be worthwhile to study this clinical question in thorough retrospective or prospective analyses to obtain ultimate information.
Whether the activation of the inflammatory process suggested here to occur as a consequence of joint destruction might be mainly due to bone damage, cartilage damage or both is worth further consideration, since cartilage and bone damage may be uncoupled. This has been elegantly shown in experimental models of arthritis,49 but we have also recently provided evidence in this respect in human RA, showing joint space narrowing and erosions may run different courses.50 All these aspects increase the level of complexity inherent to the presented hypothesis.
Further, there is a striking variability in joint damage progression seen in patients with RA, ranging from very mild to rapidly destructive and disabling disease. The differences between these extremes are far beyond a linear relationship; rather, for the disease process to generate such a broad width of destruction, an exponential relationship would be necessary. Biologically, exponential relationships are mostly a consequence of a positive feedback mechanism. However, the induction of damage by disease activity and the induction of disease activity by damage would constitute such a positive feedback loop. Autoimmune events are likely part of this game, but there are probably other factors that come into play and cause the considerable heterogeneity of the disease potentially via facilitating or engaging the presumed feedback loop of damage on the inflammatory processes.
We are aware that the hypothesis brought forward here is not in line with the mainstream of current thinking that inflammation is the major driving force of RA. We also have no reason to challenge this belief to which work from so many groups, including our own, have contributed. Rather, we wish to complement this traditional view by looking at the RA process from an additional angle.
Importantly, however, to prove this hypothesis one would need to find support in experimental models of arthritis, showing for example that microsurgical elimination of cartilage prevents the occurrence of collagen arthritis or other forms of experimental inflammatory joint disease. In addition, one would need to look for verification in analyses of clinical data from observational or controlled studies. To this end one could test (1) if the relationship between joint damage and disease activity might be similar to or even exceed that of disease activity with damage, and (2) if both these associations (activity>damage vs damage>activity) are independent of each other. Proof of this hypothesis would not change our current search for even better remedies for RA, but it might shift our focus more toward stronger interference with autoimmunity and joint damage than we may currently aim for.
REFERENCES
Footnotes
Competing interests: None declared.