Article Text

Abstract
Objective: To study the role of Toll-like receptor (TLR)2 and 4 in the onset of joint inflammation and cartilage destruction during immune complex-mediated arthritis (ICA), and its relationship with FcγR expression.
Materials and methods: ICA was induced in knee joints of TLR2−/− and TLR4−/− mice and their wild-type controls. Joint inflammation and cartilage destruction were measured in the knee joint using histology. mRNA levels were determined in synovial specimens and macrophages using quantitative polymerase chain reaction and cytokine protein levels in synovial washouts using Bioplex.
Results: Joint inflammation and cartilage destruction were not different in arthritic TLR2−/− and wild-type mice. By contrast, at day 1 after ICA induction, joint swelling and proteoglycan depletion in knee joints of TLR4−/− mice were considerably lower (inflammation 68–79% and proteoglycan depletion 27–76%) when compared with wild-type controls. Cytokine production at this time point was markedly reduced in TLR4−/− mice (interleukin (IL)1, IL6, macrophage inflammatory chemokine (MIP)-1α and keratinocyte-derived chemokine 49%, 72%, 68% and 84%, respectively). In arthritic synovia of TLR4−/− mice, and also after injection of the antigen poly-l-lysine (PLL) lysozyme alone, mRNA levels of FcγR, and the FcγR regulating cytokine IL10 were considerably lower. Stimulation of peritoneal macrophages with PLL lysozyme up regulated mRNA levels of FcγR and IL10, whereas neutralisation by anti-IL10 antibodies largely blocked FcγR up regulation. At day 4, joint inflammation and cartilage destruction were comparable in TLR4−/− mice and wild-type controls.
Conclusion: TLR4 regulates early onset of joint inflammation and cartilage destruction during ICA arthritis by up regulation of FcγR expression and enhanced cytokine production. TLR4-mediated up regulation of FcγR is largely mediated by IL10.
- FcγR, Fcγ receptors
- GAPDH, glyceraldehyde 3-phosphate dehydrogenase
- ICA, immune complex-mediated arthritis
- IFN, interferon
- LPS, lipopolysaccharides
- MIP, macrophage inflammatory chemokine
- MMP, matrix metalloproteinase
- PLL, poly-l-lysine
- TLR, Toll-like receptor
Statistics from Altmetric.com
- FcγR, Fcγ receptors
- GAPDH, glyceraldehyde 3-phosphate dehydrogenase
- ICA, immune complex-mediated arthritis
- IFN, interferon
- LPS, lipopolysaccharides
- MIP, macrophage inflammatory chemokine
- MMP, matrix metalloproteinase
- PLL, poly-l-lysine
- TLR, Toll-like receptor
IgG containing immune complexes are important triggers involved in joint inflammation and severe cartilage destruction during experimental arthritis.1–3 Comparing various experimental arthritis models, we previously found that severe cartilage destruction was observed only in those models in which immune complexes were present.4 IgG containing immune complexes present in the joint are recognised by Fcγ receptors (FcγR) which belong to the immunoglobulin superfamily.5,6 In naive joints, these FcγR are mainly expressed on lining macrophages which cover the inside of diarthrodial joints.7 We found that lining macrophages and their FcγR are of crucial importance in onset and prolongation of joint inflammation and cartilage destruction during passively induced immune complex arthritis.1,8,9 We thereby observed that macrophage depletion before arthritis induction almost completely inhibited joint inflammation.8 Using FcγR knockout mice, we additionally found that FcγRIII was the dominant FcγR regulating joint inflammation, whereas FcγRI was a more dominant receptor involved in regulating chondrocyte death.10,11 Normally, FcγR expression in naive joints is low, which suggests that during the first phase, inflammatory triggers may induce up regulation of FcγR crucial for development of arthritis.
In our immune complex arthritis model, we make use of a cationic antigen lysozyme which is coupled to poly-l-lysine (PLL) lysozyme. Due to electrical charge, this antigen binds firmly to cell surfaces and strongly increases the retention of the antigen and thereby immune complexes within the joint. In earlier studies, we found that our antigen induces an “irritans” effect of its own, which is important in the induction of the arthritis.12
The antigen itself may bind to cell membranes, thereby activating receptors whose signalling may potentiate expression of FcγR. One of the most common receptor families involved in first binding of antigens are Toll-like receptors (TLRs), which recognise a whole plethora of pathogens sharing the same pathogen-associated molecular patterns.13,14 During rheumatoid arthritis, TLRs, mainly TLR2 and 4, are strongly up regulated in the synovial layer15 and have been thought to be involved in the pathogenesis of arthritis.16 Binding of external antigens, and endogenous ligands to TLRs leads to intracellular signalling, and subsequently release of cytokines and enzymes may drive onset and prolongation of arthritis and cartilage destruction.17
In the present study, we investigated the role of TLRs and its relationship with FcγR expression in cationic immune complex arthritis using TLR2 and TLR4−/− mice. We found that TLR4 but not TLR2 is involved in early onset of joint inflammation and cartilage destruction during immune complex-mediated arthritis (ICA). This might be caused by TLR4-dependent up regulation of FcγR and production of cytokines. IL10 seemed to be a crucial intermediate in TLR4-mediated FcγR up regulation.
MATERIALS AND METHODS
Animals
TLR4−/− mice (C3H/hey mice, lacking functional TLR4)18 and C3H/hen (wild-type controls) were obtained from Jackson Laboratories, Bar Harbour, USA. TLR2−/− mice were obtained from S Akira (Osaka University, Osaka, Japan). C57BL/6 mice were obtained from Charles River WIGA Gmbh (Sulzfeld, Germany). TLR−/− and their wild-type controls (10–12 weeks old) were used in the experiments. Mice were fed a standard diet and tap water ad libitum.
Induction of ICA
ICA was passively induced by injecting 3 μg PLL lysozyme (in 6 μl phosphate-buffered saline) in knee joints of mice that had previously (16 h earlier) received intravenously polyclonal antibodies directed against lysozyme. The polyclonal antibodies were raised in rabbits. To block lipopolysaccharides (LPS), 10 μg/ml polymyxin B sulphate (Sigma-Aldrich, St Louis, Missouri, USA) was added to 3 μg PLL lysozyme before injection.
99mTc uptake measurements
Joint inflammation was measured by 99mTc pertechnetate uptake in the knee joint. Briefly, mice were injected intraperitoneally with 12 μCi 99mTc. After 30 min, γ radiation was assessed by use of a collimated Na–I-scintillation crystal with the knee in a fized position. Arthritis was scored as a ratio of the 99mTc uptake in the right inflamed (R) and left non-inflamed (L) knee joint. R:L ratios >1.1 were taken to indicate inflammation of the right knee joint.
Histology of arthritic knee joints
Total knee joints of mice were isolated 1 and 4 days after onset of arthritis. Joints were decalcified, dehydrated and embedded in paraffin wax. Tissue sections (7 μm) were stained with either haematoxylin and eosin or safranin-O. Histopathological changes were scored using the following parameters.
Inflammation was graded on a scale from 0 (no inflammation) to 3 (maximal inflamed joint) as influx of inflammatory cells in synovium (infiltrate) and joint cavity (exudate).
Several parameters of cartilage destruction were determined like proteoglycan depletion, chondrocyte death and matrix metalloproteinase (MMP)-mediated cartilage destruction. Various cartilage layers in the knee joints were measured (patella, medial tibia, medial femur, lateral tibia, lateral femur).
Proteoglycan depletion was measured as loss of red staining after staining with safranin-O, and was expressed as a percentage of fully stained total cartilage layer.
Chondrocyte death was scored as the amount of empty lacunae, expressed as the percentage of total amount of cells within the cartilage layers.
MMP-mediated cartilage destruction was measured by determining VDIPEN neoepitopes using immunolocalisation. Sections were digested with proteinase-free chondroitinase ABC (0.25 U/ml in 0.1 M Tris-HCl, pH 8; Sigma, Zwijndrecht, The Netherlands) to remove the side chains of proteoglycans, followed by incubation with affinity-purified rabbit anti-VDIPEN IgG.19 The primary antibody was detected using biotinylated goat anti-rabbit IgG and avidin–streptavidin-peroxidase (Elite kit; Vector, Burlingame, California, USA). Counterstaining was done with orange G (2%). Areas of immunostaining were expressed as percentage of the total cartilage surface. VDIPEN expression was scored as the amount of VDIPEN staining, expressed as a percentage of the total cartilage surface.4
Macrophage isolation
Mice were killed using ether vapour to prevent intraperitoneal bleeding. In all, 10 ml of ice-cold Dulbecco’s modified Eagle’s medium, 10% fetal calf serum and 1% pyruvate were injected intraperitoneally. Using a thick needle (1.1 mm), the medium containing peritoneal cells was recollected and kept on ice. In addition, peritoneal cells were adhered to plastic flasks for 1.5 h and, subsequently, attached macrophages were harvested by scraping the plastic surface using a rubber policeman. Cells were placed in 24-well plates (Costar, Acton, Massachusetts, USA), 1 ml/well, at a concentration of 2×106 cells/ml. After a 4-day adjustment period, PLL lysozyme (1 μg) was added to the culture medium. The culture medium was isolated after 24 h.
IL10 was blocked with rat anti-murine IL10 (JES5-2A5), which was obtained from ATCC, Rockville, Maryland, USA. Anti-IL10 (0.5 mg neutralised 25 000 U MIL10 in the D36 bioassay) 100 μg antibodies were added per millilitre medium of culture medium to neutralise IL10.
Quantitative detection of FcγRs and cytokine mRNA in mouse synovia using quantitative reverse transcriptase-polymerase chain reaction
Specific mRNA levels for FcγRI, II and III, IL10 and interferon (IFN)γ were detected using the ABI/PRISM 7000 Sequence Detection System (ABI/PE, Foster City, California, USA). Briefly, 1 μg of synovial or macrophage RNA was used for reverse transcriptase-polymerase chain reaction. mRNA was reverse transcribed to cDNA using oligo dT primers, and 1/20 of the cDNA was used in one PCR amplification. PCR was performed in SYBR Green Master Mix using the following amplification protocol: 2 min at 50°C followed by 40 cycles of 15 s at 95°C and 1 min at 60°C with data collection in the last 30 s. Message for murine glyceraldehyde 3-phosphate dehydrogenase (GAPDH), FcγRI, II and III, or cytokines IL10 and IFNγ was amplified using specific primers (Biolegio, Malden, The Netherlands) at a final concentration of 300 nmol/l. FcγRI: forward (5′→3′) ACA CAA TGG TTT ATC AAC GGA ACA, reverse (5′→3′) TGG CCT CTG GGA TGC TAT AAC T; FcγRII: forward GAC AGC CGT GCT AAA TCT TGC T, reverse GTG TCA CCG TGT CTT CCT TGA G; FcγRIII: forward GAC AGG CAG AGT GCA GCT CTT, reverse TGT CTT CCT TGA GCA CCT GGA T; IL10: forward ATT TGA ATT CCC TGG GTG AGA A, reverse ACA CCT TGG TCT TGG AGC TTA TTA A; IFNγ: forward CTT CTT GGA TAT CTG GAG GAA CTG, reverse AGA GAT AAT CTG GCT CTG CAG GAT. All primers are validated on serial dilutions of cDNA and amplification plots always had a slope between 2.9 and 3.5.
Relative quantification of the PCR signals was performed by comparing the cycle threshold value (Ct) of the FcγR and cytokine genes in the different samples after correction of the GAPDH content for each individual sample, and corrected for signals found in naive (t = 0) joints. The sizes of the PCR products were between 80 and 100 nucleotides.
Measurements of cytokines in synovial washouts
To determine levels of cytokines (IL1, IL6 and IL10) and chemokines (keratinocyte-derived chemokine and MIP-1α) in synovial washouts, synovial specimens were isolated in a standard manner20 and incubated in RPMI 1640 medium (GIBCO BRL, Breda, The Netherlands) for 1 h at room temperature. Cytokine and chemokine levels were determined using the BioPlex kits from BioRad (Hercules, California, USA) for the Luminex multianalyte system. Sensitivity of the cytokine kits varied between 5 and 10 pg/ml.
Statistical analysis
Differences between experimental groups were tested for significance using the Wilcoxon rank test. p Values <0.05 were considered significant.
RESULTS
TLR4 but not TLR2 regulates early onset of joint inflammation during ICA
To investigate the contribution of TLR2 and 4 in the onset and progression of ICA, joint swelling was measured 1, 2 and 4 days after ICA induction in TLR2−/−, TLR4−/− and their wild-type controls. In arthritic knee joints of wild-type mice, a severe swelling was found at day 1, which waned thereafter (fig 1). No differences in swelling were found in TLR2−/− mice. By contrast, in arthritic knee joints of TLR4−/− mice, joint swelling was 56% lower only at day 1, but not reduced at days 2 and 4 (fig 1).

Swelling of the knee joint was determined as R:L ratios of 99mTc uptake at various days (1, 2 and 4) after induction of immune complex-mediated arthritis (ICA) in the right knee joint of TLR2−/−, TLR4−/− mice and their wild-type controls. In the left knee joint, phosphate-buffered saline was injected. Values represent the mean (SD) of six mice. Two independent experiments were performed. Data were evaluated using the Wilcoxon rank test (*p<0.05). Note that the swelling was almost absent in knee joints of arthritic TLR4−/− mice but not of TLR2−/− mice at day 1 after arthritis induction.
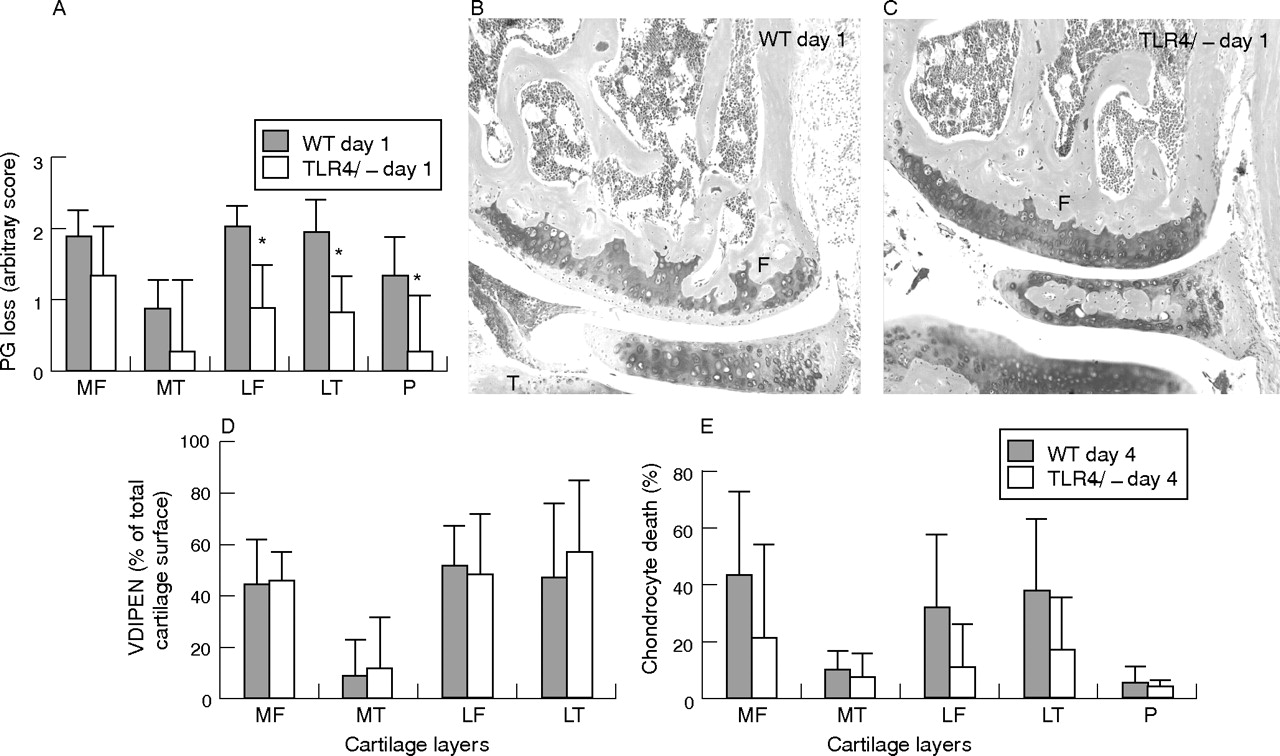
We further investigated the onset of joint inflammation by histology of total knee joint sections. The inflammatory cell mass in the cavity (exudate) and synovial layer (infiltrate) was thereby determined using an arbitrary score from 0 to 3. At day 1 after ICA induction, no differences were found in TLR2−/− mice and wild-type controls (data not shown). By contrast, joint inflammation in TLR4−/− was significantly lower (exudate and infiltrate, respectively, 85% and 65% lower) when compared with wild-type (fig 2A and photographs 2D v control 2C). At day 4 after ICA induction, inflammatory cell mass was comparable between the two strains (fig 2B), indicating that TLR4 regulates early but not late joint inflammation.

Frontal sections of whole knee joints 1 and 4 days after induction of immune complex-mediated arthritis (ICA) in TLR4−/− mice and their wild-type controls. The inflammatory cell mass present in the synovium (infiltrate) and in the knee joint cavity (exudate) was determined using an arbitrary scale from 0 to 3: 0, no cells; 1, minor; 2, moderate; 3, maximal. The inflammatory cells were counted independently by two observers blinded to the experimental set-up. Data are the mean of six mice. Two independent experiments were performed. Significance was tested using the Wilcoxon rank test (*p<0.05). Original magnification of the photographs is ×250. F, femur; P, patella; S, synovium. Note the significantly lower inflammatory mass in knee joints of TLR4−/− mice (A and photograph D v wild-type control A and photograph C) at day 1 after ICA induction. No differences were found at day 4 (B).
As joint inflammation is often correlated to cartilage destruction, we additionally investigated loss of proteoglycans. Proteoglycan depletion starts in the early phase (day 1) followed by more severe cartilage destruction like MMP-mediated matrix erosion and chondrocyte death at later phases (day 4). Proteoglycan depletion was determined as loss of red staining from various cartilage layers of the knee joint. No differences in proteoglycan depletion were found in cartilage layers of arthritic TLR2−/− mice and their controls (data not shown). By contrast, proteoglycan depletion in arthritic TLR4−/− mice was inhibited in the cartilage layers of the lateral femur, lateral tibia and patella (between 27% and 76%; fig 3A and photograph B v control C). At day 4 after arthritis induction, severe cartilage destruction (chondrocyte death and MMP-mediated matrix destruction (VDIPEN)) had developed. MMP-mediated cartilage destruction data were not significantly different, while chondrocyte death data, despite the values being somewhat lower in TLR4−/− mice, did not reach significance (fig 3D, E).

Measurement of cartilage destruction: proteoglycan depletion (A–C) at day 1 and matrix metalloproteinase (MMP)-mediated cartilage destruction (expression of VDIPEN neoepitopes) (D) and chondrocyte death (E) at day 4 after ICA induction in knee joints of TLR4−/− and their wild-type controls. Proteoglycan depletion was determined in various cartilage layers of the knee joint (P, patella; MF, medial femur; MT, medial tibia; LF, lateral femur; LT, lateral tibia) and was significantly lower in arthritic knee joints of TLR4−/− at day 1 after arthritis (A and photographs C v control B). F, femur; M, meniscus and T, tibia. VDIPEN staining was measured at day 4 and expressed as percentage positive staining of the total cartilage area. No significant differences were found between TLR4−/− and their wild-type controls at day 4 after ICA induction. Chondrocyte death was expressed as percentage of empty lacunae of the total cartilage area. Note that chondrocyte death was lower in TLR4−/− mice, although values did not reach significance (E). Data represent the mean (SD) of seven mice and were statistically evaluated using the Wilcoxon rank test. *p<0.05.
TLR4 stimulates cytokine production during ICA
To further investigate the involvement of TLR4 in early onset of joint inflammation and proteoglycan depletion, we additionally measured cytokine concentrations in washouts of synovial specimen using Bioplex. Protein levels of MIP-1α, keratinocyte-derived chemokine, IL1 and IL6, crucial cytokines involved in inflammation and proteoglycan breakdown, were high in washouts of day 1 arthritic wild-type joints (172, 69, 149 and 2121 pg/ml, respectively; table 1). By contrast, in washouts of day 1 arthritic TLR4−/− mice, cytokine protein levels were significantly lower (49%, 72%, 68% and 84%, respectively).
Release of various cytokines (macrophage inflammatory chemokine-1α, keratinocyte-derived chemokine, interleukin (IL)1 and IL6) 1 day after induction of immune complex-mediated arthritis in knee joints of TLR4−/− and their wild-type controls
TLR4 is related to potentiation of FcγR in the arthritic knee joint
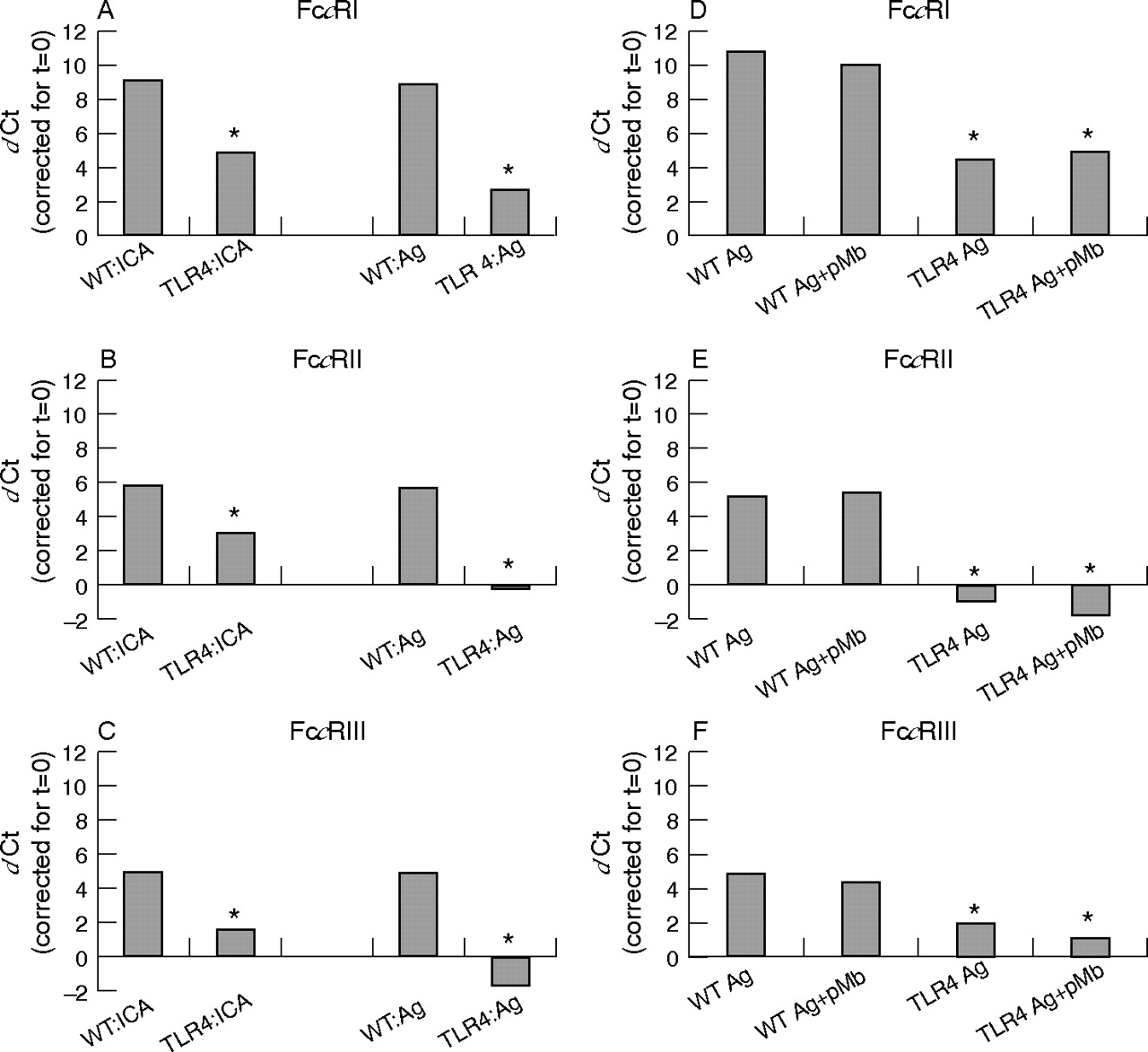
As joint inflammation between TLR4−/− and wild-type controls was different only at day 1 and as the onset and prolongation of ICA is mediated by FcγR,1 as shown earlier in our laboratory, we additionally investigated whether TLR4 mediates FcγR expression within this model. At 1 day after ICA induction, mRNA levels of FcγR (particularly FcγRI) became highly increased in the synovial layer of wild-type controls (FcγRI, II and III was raised 512, 32 and 32 times, respectively). When ICA was induced in the knee joints of TLR4−/− mice, mRNA levels of FcγR were significantly lower (FcγRI, II and III 47%, 48% and 69%; fig 4A–C) when compared with arthritic controls.

Expression of mRNA levels of FcγR (FcγRI, II, III) in inflamed synovia of TLR4−/− and their wild-type controls. Synovia were isolated 1 day after immune complex-mediated arthritis induction or injection of the antigen PLL lysozyme alone (left panel). Furthermore, the effect of LPS contamination in PLL lysozyme on synovium was studied by inhibiting LPS (3 μg/6 μl) with 10 μg/ml polymyxin B (pMb; right panel) before injection. The cycle threshold value (Ct) of the various FcγR genes was corrected for GAPDH content and t = 0. FcγR expression was similar in naive knee joints (t = 0) of TLR4−/− and controls. Note that at day 1 after ICA induction significantly lower levels of FcγR levels are measured in TLR4−/− (A–C). Injection of the antigen (Ag) alone also induced high expression of FcγR in wild-type knee joints not different from ICA. In TLR4−/−, FcγR expression, was almost absent. Note that LPS blockade by polymyxin B (Ag+pMb) did not alter FcγR expression, indicating that the physicochemical properties of PLL lysozyme determine TLR4 stimulation (D–F). Data represent the mean (SD) of two independent experiments. In each experiment, seven mice were used. Values were statistically evaluated using the Wilcoxon rank test (significance *p<0.05).
Intra-articular injection of the antigen alone (3 μg PLL lysozyme) also potentiated FcγR mRNA expression within the synovium not different from that found after early ICA induction (at day 1, FcγRI, II, III, 512, 32 and 32 times, respectively). In TLR4−/− mice, however, FcγR expression was almost completely blocked (FcγRI, II and III lowered by 70%, 100% and 100%, respectively; fig 4A–C).
One of the explanations why PLL lysozyme potentiates FcγR expression via TLR4 might be contamination by LPS. A small amount of LPS (10 pg) was detected in 6 μl PLL lysozyme using a Lal test. However, when this LPS was neutralised by adding a surplus of polymyxin B to PLL lysozyme just before intra-articular injection, comparable potentiation of FcγR mRNA expression was found within the synovium of wild-type mice, whereas FcγR expression was again significantly lower in TLR4−/− at 24 h after injection (fig 4D–F). Polymixin B alone had no effect on FcγR expression (data not shown). Moreover, intra-articular injection of 10 pg LPS also did not increase FcγR expression (data not shown). These data suggest that physicochemical characteristics (high cationicity) of the antigen rather than LPS contamination is responsible for TLR4 triggering.
TLR4 potentiates FcγR expression by production of IL10
To further investigate how PLL lysozyme mediates FcγR up regulation via TLR4, we additionally investigated whether PLL lysozyme may act by stimulating synovial cells to release cytokines like IFNγ and IL10, which have been shown to up regulate FcγR. Injection of PLL lysozyme (together with polymixin B) into knee joints of wild-type control mice significantly increased mRNA levels of IL10 (4 times; fig 5A) but not IFNγ in the synovial layer. In TLR4−/− synovia, IL10 mRNA levels were 72% lower. In line with this, IL10 protein levels in synovial washouts of PLL lysozyme-injected TLR4−/− mice were 63% lower than in washouts derived from wild-type knee joints (fig 5B).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
IL10 mRNA and protein levels were determined in synovia of knee joints, 1 day after injection with 3 μg PLL lysozyme. Protein levels were measured using Bioplex (sensitivity 10 pg/ml). IL10 mRNA (A) and protein (B) levels were significantly lower in arthritic synovia or washouts of TLR4−/− when compared with controls. Neutralisation of IL10 using anti-IL10 antibodies during stimulation of peritoneal macrophages by PLL-lysozyme inhibited FcγR (FcγRI, II, III) up regulation (C). Data represent the mean (SD) of two independent experiments. In each experiment, seven mice were used. Data were statistically evaluated using the Wilcoxon rank test (*p<0.05).
As macrophages are crucial cells in the knee joint capable of IL10 production, we additionally investigated whether IL10 may be involved in regulating PLL lysozyme-regulated FcγR expression in macrophages. Murine macrophages were stimulated with 1 μg of PLL lysozyme in the presence of 100 μg of anti-IL10 antibodies (Jes5-2A5) or isotype-specific control antibodies. PLL lysozyme up regulated FcγRI, II or III mRNA levels. Anti-IL10 antibodies completely blocked FcγRI up regulation and concomitantly also down regulated expression of FcγRII and III (FcγRI, II and III, 32, 32 and 32 times, respectively; fig 5C).
DISCUSSION
In the present study, we found that TLR4 but not TLR2 is important in driving early onset of joint inflammation and cartilage destruction during ICA. In previous studies, we have shown that FcγR expressed on synovial macrophages are crucial in mediating the onset of ICA.1 However, in a naive knee joint, FcγR are expressed in only low amounts on lining macrophages. Activation of TLR4 by the antigen alone led to strong potentiation of FcγR. FcγRII and III up regulation seemed thereby strongly TLR4 dependent. Injection of the antigen along with polymixin-B, an LPS neutralising agent, further stimulated FcγR expression. The latter indicates that it is not LPS but the physicochemical characteristics of the antigen that are involved in FcγR potentiation. PLL has been shown to strongly stimulate monocytes.21 PLL lysozyme is highly cationic and, owing to its electric charge, firmly binds to the negatively charged glycocalix on the surface of synovial lining cells. Cellular stress leads to increased production of heat shock proteins (like HSP60 and 70), which have been associated with arthritis.22,23 In the present study, direct stimulation of macrophages by HSP60 did not up regulate FcγR expression, suggesting that HSP60 is not the factor mediating TLR4 activation (data not shown). Membrane binding of PLL lysozyme may, however, lead to release of other endogenous ligands like fibrinogen,24 surfactant protein A,25 fibronectin extra domain26 or soluble hyaluronan,27 all of which have been shown to activate TLR4 signalling. The latter may lead to activation of nuclear factor-κB and interferon regulatory factor 3, and release of cytokines and chemokines19 involved in FcγR regulation. Two major cytokines involved in FcγR regulation are IFNγ and IL10. Both cytokines increase the FcγR balance in favour of activating FcγR, and are associated with a strong capacity of monocytes to respond to IgG-triggered tumour necrosis factor α production.28 Although IFNγ is produced by macrophages,29 no up regulation of IFNγ mRNA was found in the synovial layer after intra-articular injection of PLL lysozyme. By contrast, IL10 mRNA and protein levels became up regulated in wild-type synovia and seemed significantly lower in TLR4−/−. Moreover, inhibition of IL10 blocked up regulation of FcγR. This is in line with earlier studies which showed that stimulation of TLR4 caused up regulation of IL1030 and that IL10 stimulated macrophage functions including up regulation of predominantly FcγRI.31 Up regulation of FcγR by IL10 may lead to greater vulnerability of macrophages towards activation by immune complexes. Moreover, binding of IC to FcγR also resulted in up regulation of IL10 by macrophages,32 thereby further amplifying FcγR expression and cell activation.
Up regulation of FcγR expression by TLR4 signalling up regulated not only IL10 but also other proinflammatory cytokines like IL1 and IL6. IL1 is a dominant cytokine involved in joint inflammation and cartilage destruction during ICA.32 Earlier studies showed that neutralisation of IL1 by anti-IL1 antibodies or IL1ra largely blocked synovial inflammation and cartilage destruction.32,33 IL1 probably acts by promoting synovial cells to secrete chemokines like keratinocyte-derived chemokine and MIP-1α. The latter chemokines are dominant attractants for, respectively, PMN and monocytes.34 Inhibition of IL1 and subsequently release of keratinocyte-derived chemokine and MIP-1α in TLR4−/− mice may explain why onset of early synovitis and proteoglycan depletion is inhibited.
The present study shows that TLR4 but not TLR2 can act as an early regulator of induction of activatory FcγR, which is largely mediated by IL10. TLR4 signalling provides initial stimulation setting the threshold for immune complex-driven FcγR activation. The regulatory role of TLR4 may enable the immune system to respond more efficiently to foreign antigen bound within immune complexes that are potentially harmful for the host.
REFERENCES
Footnotes
-
Published Online First 20 October 2006
-
Funding: This work was supported by the Dutch Arthritis Association.
-
Competing interests: None declared.