Article Text

Abstract
Maintenance of oxygen homoeostasis is the basic principle in cell proliferation, differentiation, survival, and function in all higher organisms. The transcription factor, HIF (hypoxia inducible factor) has a central role in oxygen homoeostasis, and is indispensably linked to energy metabolism. Abnormally reduced oxygen concentrations leading to dysfunctional cell metabolism are found in rheumatoid arthritis and hence, knowledge of the molecular adaptive responses to hypoxia and the involvement of HIF in the pathogenesis of RA are interesting.
- AIA, adjuvant induced arthritis
- ARNT, aryl hydrocarbon receptor nuclear translocator
- bHLH, basic helix-loop-helix
- CIA, collagen induced arthritis
- COX-2, cyclo-oxygenase-2
- CTAD, C-terminal transactivation domain
- CTL, cytotoxic T lymphocyte
- EC, endothelial cells
- EPO, erythropoietin
- FIH, factor inhibiting HIF-1
- HIF, hypoxia inducible factor
- HSP, heat shock protein
- IL, interleukin
- MAPK, mitogen activated protein kinase
- MMP, matrix metalloproteinase
- ODD, oxygen dependent degradation domain
- PHD, prolyl hydroxylase domain-containing protein
- PI3K, phosphatidylinositol 3-kinase
- pVHL, von Hippel-Lindau tumour suppressor protein
- RA, rheumatoid arthritis
- ROS/RNS, reactive oxygen and nitrogen species
- SDF-1, stromal cell derived factor 1
- TNFα, tumour necrosis factor α
- VEGF, vascular endothelial growth factor
- energy metabolism
- hypoxia
- hypoxia inducible factor
- oxygen homoeostasis
- rheumatoid arthritis
Statistics from Altmetric.com
- AIA, adjuvant induced arthritis
- ARNT, aryl hydrocarbon receptor nuclear translocator
- bHLH, basic helix-loop-helix
- CIA, collagen induced arthritis
- COX-2, cyclo-oxygenase-2
- CTAD, C-terminal transactivation domain
- CTL, cytotoxic T lymphocyte
- EC, endothelial cells
- EPO, erythropoietin
- FIH, factor inhibiting HIF-1
- HIF, hypoxia inducible factor
- HSP, heat shock protein
- IL, interleukin
- MAPK, mitogen activated protein kinase
- MMP, matrix metalloproteinase
- ODD, oxygen dependent degradation domain
- PHD, prolyl hydroxylase domain-containing protein
- PI3K, phosphatidylinositol 3-kinase
- pVHL, von Hippel-Lindau tumour suppressor protein
- RA, rheumatoid arthritis
- ROS/RNS, reactive oxygen and nitrogen species
- SDF-1, stromal cell derived factor 1
- TNFα, tumour necrosis factor α
- VEGF, vascular endothelial growth factor
The pathogenesis of rheumatoid arthritis (RA) involves a symmetric polyarticular arthritis that primarily affects the small diarthrodial joints of the hands and feet and, finally, progressively destroys these and other joints (reviewed by Firestein1). In the inflamed RA joint the synovium is highly infiltrated by CD4+ T cells, B cells, and macrophages, and the intimal lining becomes hyperplastic owing to the increased number of macrophage-like and fibroblast-like synoviocytes (tissue swelling/tumour). This hyperplastic intimal synovial lining forms an aggressive front, called pannus, which invades cartilage and bone structures, leading to the painful (dolor) destruction and compromised function of affected joints (functio laesa). The distance between blood vessels and synoviocytes is increased by hyperplasia of the synovial lining. This microenvironmental alteration causes hypoxia and hypoperfusion because the oxygen diffusion limit is exceeded (>100–200 μm). In addition, proliferation of synoviocytes, accumulation of infiltrates, and a wounded or incapacitated vascular network contribute to an increase in the demand for oxygen. Hypoxia and hypoperfusion, in turn, induce both an engorgement of the capillaries that carry the blood away and an increase in the diameter of the vessels that deliver blood. This results in erythema (rubor) and tissue heat (calor) and leads to the induction of angiogenesis, which ensures a constant supply of cytokines, growth factors, and infiltrating effector cells, perpetuating inflammation, infiltration, and hyperplasia. Therefore, development and persistence of RA could be supported by a progressive but dysregulated angiogenesis that leads to low levels of oxygen and nutrients in the inflamed joint.
THE RA SYNOVIUM IS HYPOXIC
Indeed, microenvironmental conditions in the inflamed joint are characterised by low partial pressure of oxygen. In 1970, the first data supporting the hypoxic nature of the RA synovium were achieved by measuring oxygen tension in samples of synovial fluids of patients with RA with a Clark-type electrode.2,3 Other studies supported these observations by reporting decreased oxygen and glucose levels and raised carbon dioxide, lactate, and acetate levels, which lead to a decreased synovial pH level (acidosis) as a result of an anaerobic metabolism.4,5 In addition, these data were confirmed by nuclear magnetic resonance spectroscopy, which displayed a profile of low molecular weight metabolites, consistent with hypoxia.6 Today, pimonidazole (Hypoxyprobe-1; Chemicon Europe, Chandlers Ford, UK), a compound, which binds to cells with low oxygen tension, is widely used to detect hypoxic areas. This compound forms protein adducts in hypoxic cells, detectable by antibodies. This method was used by Peters et al to show hypoxic areas, including structures such as the synovium, pannus, bone marrow, and articular cartilage chondrocytes in an adjuvant induced arthritis (AIA) rat model.7
“Hypoxic areas can be detected by pimonidazole”
Thus, an acute need arises for all affected cells in these hypoxic areas to:
-
Promote the cellular transition from oxidative phosphorylation to anaerobic glycolytic metabolism
-
Stimulate local angiogenesis and vascularisation
-
Raise systemic erythropoiesis in order to escape oxygen deprivation.
During the past decade the pivotal molecular mechanisms in cellular adaptive response to hypoxia have been elucidated in detail. One principal regulator of this adaptive response to hypoxia is assigned to the transcription factor, HIF (hypoxia inducible factor)8. It has been demonstrated that this factor is inducible by hypoxia and by other physical (mechanical stress, heat, and low pH) and biochemical factors (cytokines, hormones, growth factors).9–20
In addition to the staining using Hypoxyprobe-1, Peters et al identified the expression of HIF in an AIA model. In human cells of the inflamed joint (synovial sections) HIF is detectable in affected cell types, such as synovial macrophages, fibroblasts, and CD3+ T cell infiltrates, in contrast with healthy controls.21–24 In 2003, Cramer et al provided fundamental functional evidence for HIF in models of acute and chronic inflammation. In these models, deletion of HIF in macrophages and neutrophils resulted in a complete loss of the inflammatory response.25
Together, these findings indicate that HIF is involved in the persistence of inflammation and progression of neovascularisation during RA. This review summarises current knowledge about the structure and regulation of HIF-1 and focuses on its implication in RA, providing new potential therapeutic targets.
HIF: STRUCTURE AND REGULATION
HIF structure
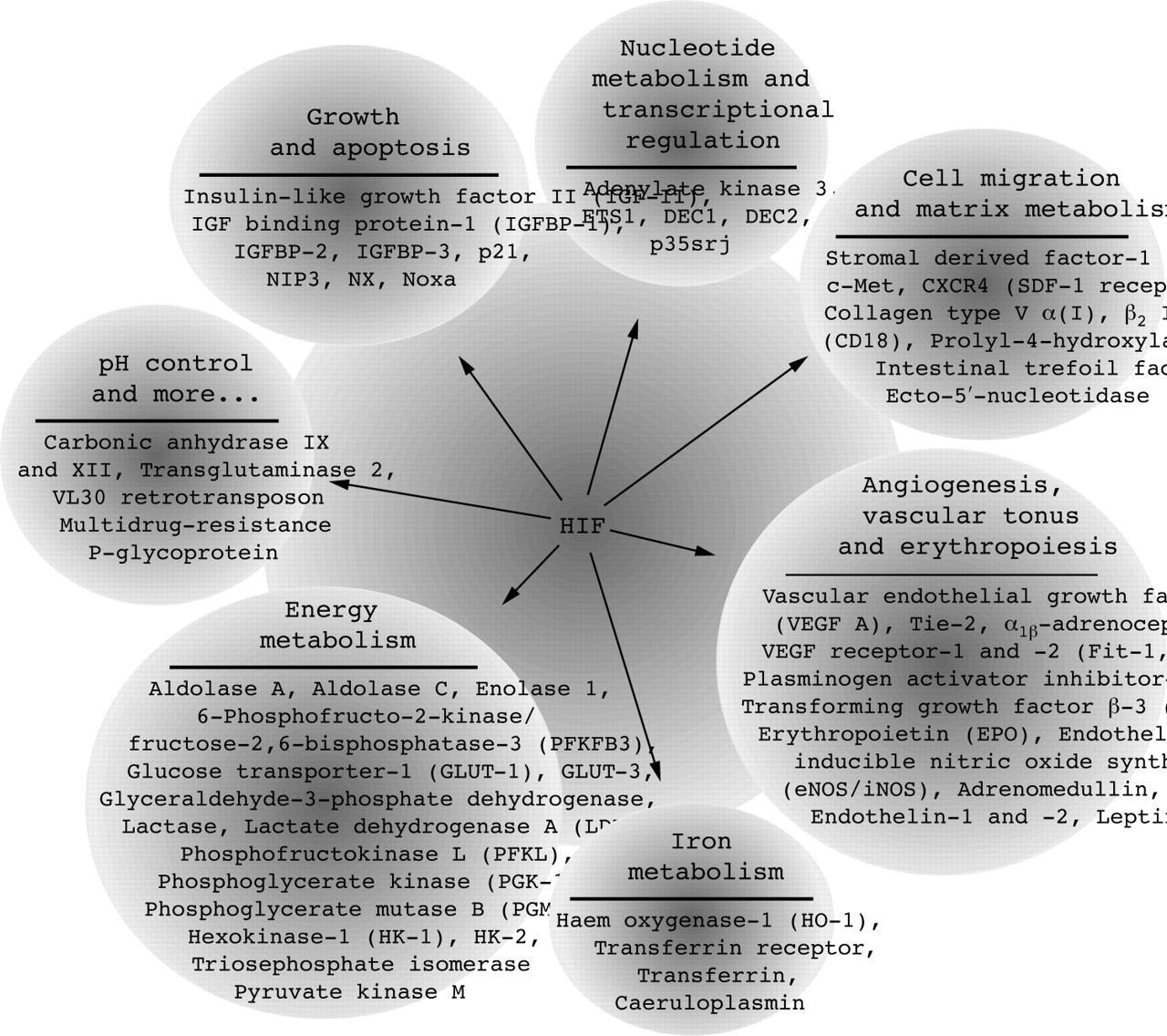
The heterodimeric transcription factor HIF is composed of two basic helix-loop-helix (bHLH) proteins—HIFα and HIFβ. Both are members of a superfamily of proteins containing a PAS domain, defined by sequence homology to the Drosophila proteins Per and Sim and the mammalian proteins AHR and ARNT.26,27 HIF subunits and binding partners are classified as the bHLH-PAS family, according to the N-terminal located domains, which are essential for DNA binding (bHLH) and nuclear dimerisation (PAS).27 The HIFα/β dimer binds to a core DNA motif (5′-G/ACGTG-3′) in the hypoxia responsive elements, which are associated with a broad range of target genes, such as vascular endothelial growth factor (VEGF), glucose transporter-1, and erythropoietin (EPO) (see fig 1). Thus, induction of HIF-DNA binding promotes erythropoiesis, angiogenesis, vasodilatation, cell growth and apoptosis, cell migration, and a switch to a glycolytic cell metabolism.

Transcriptionally influenced direct targets of HIF. Gene products, which are directly activated by HIF at the transcriptional level, are shown. So far more than 60 HIF target genes have been identified by binding of HIF to hypoxia responsive elements in association with transcriptional control sequences. These HIF target genes promote erythropoiesis, angiogenesis, vasodilatation, and a switch to a glycolytic cell metabolism, resolving and counteracting hypoxic conditions.
Whereas HIFβ (also known as aryl hydrocarbon receptor nuclear translocator, ARNT) is constitutively expressed, the levels of HIFα are highly inducible by hypoxia. Three related HIFα subunits that share structural similarities are encoded by distinct gene loci. The first and best characterised subunit is HIF-1α. HIF-1α and HIF-2α (also known as EPAS-1) have a similar gene and protein structures, are both ubiquitously expressed, and are regulated similarly.8,27–31 HIF-3α is less closely related and has been associated with a negative regulation mechanism of HIF induced gene transcription.32–35 Nevertheless, regulation, function, and tissue specific expression of HIF-3α still have to be elucidated.
“HIFα is highly inducible by hypoxia”
In addition to the bHLH-PAS domain, HIF-1α and HIF-2α share a similar oxygen dependent degradation domain (ODD) of approximately 200 amino acids in the central region of the molecule.36 The ODD—essential for the proteolytic regulation of HIF—overlaps with the internal N-terminal transactivation domain. A second transactivation domain is located at the C-terminus of the HIFα subunit (CTAD).37 CTAD confers the oxygen regulated interaction with general transcriptional coactivators, like p300/CREB binding protein and p160/steroid receptor coactivator-1, and enables the transcriptional activation of target gene expression (fig 2). These coactivators possess histone acetyltransferase activity and can remodel the chromatin structure to activate gene transcription in general.38,39

Domain structure of HIF-1α and interacting factors. Domains: NLS, nuclear localisation signal; bHLH, basic helix-loop-helix (DNA binding and dimerisation); PAS, Per/Ahr-ARNT/Sim (dimerisation); PAC, PAS associated C-terminal domain; NTAD, N-terminal transactivation domain (transcriptional transactivation); CTAD, C-terminal transactivation domain (transcriptional transactivation); Factors: PHD, HIF prolyl hydroxylase; ARD1, ADP-ribosylation factor domain; FIH, factor inhibiting HIF; pVHL, von Hippel-Lindau tumour suppressor protein.
Six transcript variants/isoforms of HIF-1α, one for HIF-2α, and six for HIF-3α have been identified (fig 3).28–31,40–45 Some of these isoforms lack parts of the ODD and the CTAD but can dimerise with ARNT or HIF-1α. In this case, dimerisation leads to transcriptionally inactive heterodimers underlining a post-transcriptional regulatory mechanism, which inhibits the HIF target gene expression. One of these shortened isoforms is the human HIF-3α2, also known as inhibitory PAS protein.33–35

Structural alignment of human HIF-1/2/3α subunits and their isoforms. Functional domains are illustrated as boxes of various shades: bHLH, basic helix-loop-helix; PAS, Per/Ahr-ARNT/Sim; PAC, PAS associated C-terminal domain; NTAD, N-terminal transactivation domain; CTAD, C-terminal transactivation domain; ODD, oxygen dependent degradation domain. Figures in square brackets are references.
Oxygen dependent regulation of HIF: normoxia
Recent studies have shown that HIF is regulated by an unprecedented signalling mechanism at the protein level. Under normoxic conditions HIFα is targeted for rapid ubiquitination and proteasomal degradation (fig 4).

Oxygen dependent HIF regulation. HIFα targeting for proteasomal degradation under normoxic conditions (left side); hypoxic stabilisation of HIFα and recruitment of transcriptional coactivators (right side). FIH, factor inhibiting HIF-1.
HIFα targeting is achieved by hydroxylation of specific prolyl residues (Pro402 and Pro564 in the case of HIF-1α) located in the ODD and acetylation of an associated lysine (Lys532). The N-acetyltransferase ARD1 (ADP-ribosylation factor domain) mediated acetylation reaction facilitates the targeting of prolyl hydroxylation by HIF.46 This prolyl hydroxylation is catalysed by specific oxygenases, identified as prolyl hydroxylase domain-containing proteins (PHDs). Cosubstrates such as iron, 2-oxogluterate and molecular oxygen are absolutely necessary for this hydroxylation reaction. Withdrawal of a cosubstrate, by iron chelators or blockade of PHD substrate binding, by cobaltous ions, inhibits the hydroxylation reaction and leads to a subsequent stabilisation of HIFα subunits. Thus, PHDs provide a direct link between oxygen sensing and stabilisation of HIFα (reviewed by Schofield and Ratcliffe, 47 Yuan et al48).
Another component of the degradation process, the von Hippel-Lindau tumour suppressor protein (pVHL), recognises hydroxylated HIFα subunits. pVHL is part of a multiprotein ubiquitin ligase complex (pVHL-E3-ligase complex) and facilitates degradation of HIFα through the ubiquitin-proteasome pathway.49 Loss of function of pVHL or PHD as well as the already mentioned inhibition of the hydroxylation reaction leads to a constitutively stabilised HIFα and an activated HIF transactivation.25,50,51
Oxygen dependent regulation of HIF: hypoxia
During hypoxia, prolyl hydroxylation is suppressed, leading to the escape of HIFα subunits from pVHL mediated proteasomal degradation and to the accumulation of high levels of HIF. Once HIFα escapes pVHL dependent degradation, heat shock protein 90 (HSP90), a molecular chaperone, associates with HIFα. The displacement of the chaperone by HIF-1β facilitates the development of a transcriptionally active and stable HIF complex, leading to HIF target gene expression.52
In addition, transactivation capability/activity of HIFα is regulated by another iron, oxygen, and 2-oxogluterate dependent oxygenase with hydroxylase capacity, called factor inhibiting HIF-1 (FIH). FIH represses HIFα transcriptional activity in two ways: (a) by recruitment of histone deacetylases, which repress chromatin remodelling and (b) by hydroxylation of an asparagine residue (Asn803) located in the CTAD, which inhibits binding of general coactivators (p300/C binding protein and p160/SRC–1) to HIFα.53–58
To achieve full transactivation capability and to activate HIF target gene expression, phosphorylation of the HIF-CTAD by p42/44 mitogen activated protein kinase (MAPK) or p38MAPK is necessary.59,60 These findings were derived from the observation that MAPK inhibitors attenuated HIF-1α transactivation ability but not protein stabilisation and DNA binding activity.61,62 The essential phosphorylation site has not yet been confirmed.
Oxygen independent regulation of HIF
Beside hypoxia, several stimuli have been reported to induce HIFα stabilisation and transcriptional activity, including mechanical stress, cobaltous ions, iron chelators, reactive oxygen and nitrogen species (ROS/RNS), hormones, cytokines and growth factors, and loss of function of the tumour suppressor proteins, pVHL or PTEN (fig 5).9–20

Factors inducing HIF transactivation activity. Cytokines: interleukin 1β (IL1β), tumour necrosis factor α (TNFα); hormones: insulin, thyroid hormone, follicle stimulating hormone (FSH), angiotensin II, and thrombin; hypoxia, low pH level (acidosis), heat (fever); reactive oxygen species (ROS) and reactive nitrogen species (RNS): hydroxyl radicals (−OH*), superoxide anions (−O2*−), hydrogen peroxide (H2O2), singlet oxygen (1O2), nitric oxide (NO), nitrous acid (HNO2), and nitrogen dioxide (NO2); bacterium and virus related: lipopolysaccharide (LPS), hepatitis B virus protein X (HBx); growth factors: basic fibroblast growth factor (bFGF), epidermal growth factor (EGF), hepatocyte growth factor (HGF), insulin-like growth factor-I and -II (IGF-I/II), platelet derived growth factor-BB (PDGF-BB), and heregulin.
CoCl2, iron chelators, and loss of function mutations of tumour suppressor proteins directly affect the degradational pathway of HIFα. Hormones, proinflammatory mediators such as interleukin (IL) 1β, tumour necrosis factor α (TNFα), and nitric oxide and growth factors can induce HIF-1α through ligand binding to their receptors. Similarly, HIF induction could be observed using mechanical stress by stimulating stretch activated channels, which has obvious relevance for RA and swollen joints where there is high intra-articular pressure.63,64 Both methods activate the phosphatidylinositol 3-kinase (PI3K)/serine threonine protein kinase B (AKT)/FRAP (FKBP12-rapamycin associated-protein) signalling cascade.64–66 A second pathway via RAF/MEK/MAPK conveys transcriptional transactivation activity to the HIF α/β dimer by phosphorylation of CTAD and its coactivators. Both pathways can be directly activated by a ligand receptor interaction or through the GTP dependent adapter molecule, RAS (fig 6).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Hormone, cytokine, and growth factor mediated signalling to regulate HIF-1α protein expression and transcriptional transactivation activity. Hormones like insulin, cytokines like tumour necrosis factor (TNFα) and interleukin 1β (IL1β), as well as growth factors like transforming growth factor β (TGFβ) and fibroblast growth factor 2 (FGF2) can induce HIF-1 target genes even under normoxic conditions. Binding of these factors to their receptors activates intracellular signalling cascades by kinase mediated phosphorylation. One activated cascade is the phosphatidylinositol 3-kinase (PI3K)/AKT (protein kinase B)/FK506 binding protein12-rapamycin associated protein leading to an increased HIF-1α mRNA translation, sufficient for its stabilisation. A negative regulator of the PI3K/AKT/FRAP cascade is the tumour suppressor protein PTEN, a phosphatase, which abolishes AKT activation. A second pathway involved in HIF-1 signalling is the RAF/MEK/MAPK pathway, leading to an enhancement of transcriptional activity of HIF-1α. Both pathways can be stimulated by the small GTP binding protein RAS.
Inhibition of PI3K by wortmannin or LY294002, de-phosphorylation of AKT by overexpression of PTEN, restoration of functionally inactive PTEN, or inhibition of FRAP by rapamycin resulted in attenuation of HIF-1α stability and transcriptional activity.64–68 In detail, FRAP de-represses the translational regulatory protein and promotes the translation from the 5′UTR of HIF-1α mRNA, leading to an increase in HIF-1α protein.65
Recently, another report provided further evidence for a PI3K/Akt mediated stabilisation of HIF-1α by inducing expression of the HSP90 and HSP70. Both have been found to interact directly with HIF-1α. As described above, HSP90 chaperones HIF-1α in a ready state for dimerisation with ARNT through binding to the PAS domain of HIF-1α. In addition, HSP70 has been shown to interact directly with the ODD of HIF-1α suggesting a possible stabilisation mechanism, which has to be confirmed in future studies.69
IMMUNE RESPONSE REQUIRES FUNCTIONAL AND TIGHTLY REGULATED HIF
To analyse cellular and physiological functions of HIF-1α in more detail, several gene targeted knockout embryonic stem cell models and mice models, as well as in vitro studies using RNA interference technology, have been designed and described.24,25,70–75
Disruption of both alleles for HIF-1α in mice (HIF-1α−/−) leads to embryonic lethality owing to mesenchymal cell death, vascular regression, and abnormal remodelling in cephalic regions.72
To bypass embryonic lethality Kojima et al used the RAG-2 deficient blastocyst complementation system of HIF-1α null mice, resulting in HIF-1α−/− T and B lymphocytes. Interestingly, they found no significant changes in the proportion of T cell subsets or T cell developmental alterations, but observed functional changes in cytotoxic T lymphocytes (CTLs). Under normoxic as well as under hypoxic conditions HIF-1α −/− CTLs were much more nearly non-cytotoxic.76–78 However, mice with thymocyte-specific overexpression of HIF-1α owing to the loss of HIF degrading factor, pVHL, showed increased caspase 8 mediated apoptosis of thymocytes. Additional loss of HIF reversed thymocyte apoptosis, thus demonstrating a need for proper regulation of HIF-1 transcriptional activity through pVHL in normal thymocyte development.79
Similarly, HIF-1α−/− B lymphocytes failed to undergo normal postnatal development. This resulted in a reduced postnatal bone marrow B cell production, which was compensated by a homoeostatic proliferation of B cells (B1-like lymphocytes) of fetal origin. These accumulated peritoneal B1-like lymphocytes were found to show high expression of the B220 (CD45) receptor associated protein tyrosine phosphatase and significant autoimmune reactivity (accumulation of anti-dsDNA antibodies and rheumatoid factor in serum, deposits of IgG and IgM in kidney, and proteinuria). These findings can be interpreted as a consequence of an abnormal balance of proliferation versus cell cycle arrest controlled by HIF-1α, which in turn leads to a rheumatic disease, such as the lupus-like syndrome.77,78 Using conditional deletion of HIF-1α in CD19+ B cells, Goda et al confirmed the crucial role of HIF in the induction of cell cycle arrest by stabilisation of p53.71 p53 regulates progression through the cell cycle, and responds to DNA damage and oxidative stress/hypoxia by initiating cell cycle arrest, repair, or apoptosis. The MDM2 protein negatively regulates the transcriptional activity of p53 by binding to the p53 transactivation domain, resulting in inactivation, nuclear export, and targeting for destruction by ubiquitination. During hypoxia p53 competes with HIF-1β for dimerisation with HIF-1α. HIF-1α bound p53 forms a ternary complex with MDM2, which in turn targets HIF-1α instead of p53 for ubiquitination and proteasomal degradation, promoting p53 stability and activity.80 In contrast with tumour cells, B cells lacking HIF showed a higher proliferative activity during hypoxia.71,81
Although, HIF-1α promotes cell survival and cellular functionality, it is involved in induction of cell cycle arrest and apoptosis through activation and stabilisation of p53. p53 has also been found to have a role in the pathology of RA, as mutations in the p53 gene locus provoked by ROS/RNS fail to stop proliferation (reviewed by Tak et al82).
“Hypoxia enhances cell activation and immunological function of stimulated immune cells”
Deletion of HIF in endothelial cells (EC) leads to the interruption of an autocrine loop necessary for the hypoxic induction of VEGFR-2 expression. Thus, HIF-1α affects the function of EC during their migration to areas of hypoxia and increased levels of VEGF, indicating a necessary feedback mechanism during the hypoxic response of EC for proper angiogenesis and neovascularisation for wound healing.83
Conditional deletion of HIF-1α in myeloid cells, like neutrophils and macrophages, drastically reduces the cellular ATP pool. The metabolic defect results in profound impairment of myeloid cell aggregation, motility, invasiveness, and bacterial killing and therefore an impairment of a functional innate immune response as described by Cramer et al.25 Conversely, loss of pVHL and constitutive expression of HIF-1α leads to a large increase in acute inflammatory responses, indicating that hypoxia enhances cell activation and immunological function of stimulated immune cells. Thus, Cramer et al concluded that HIF-1α is essential for the regulation of the glycolytic capacity in myeloid cells and that myeloid cells are highly dependent on glycolytic rather than on mitochondrial ATP synthesis.
In summary, loss of HIF-1α has been shown to impair normal development in different cells and tissues via vascular regression, deregulation of the balance between proliferation and apoptosis, and loss of cellular ATP restoration during hypoxia. In addition, the effector cell function is widely impaired and the ATP pool is drastically reduced, both suggesting the importance of HIF-1α in the immune response.
IMPLICATIONS OF HIF IN RA
Enhanced expression of HIF has been found under several different pathophysiological conditions such as ischaemic and cardiovascular disorders, pulmonary hypertension, pregnancy disorders, and cancer. Up regulated HIF has also been detected in immune cells involved in acute inflammation during experimental wound healing in the rat model.84
HIFα is expressed in RA
Using immunohistochemistry, Hollander et al and Giatromanolaki et al observed an HIF-1α up regulation in synovial CD68+ macrophages prepared from biopsy samples of patients with RA and osteoarthritis in comparison with healthy controls. In addition, they also observed increased expression of HIF-1α and HIF-2α in the synovial lining and in stromal cells in both diseases.21,23
Loss of HIF-1α results in reduction of joint inflammation
As mentioned above, a milestone in the study of HIF was the finding of Cramer et al that HIF-1α is essential for myeloid cell mediated inflammation. Using a passively induced arthritis model, they provided the first evidence of the role of HIF-1 in joint inflammation. Deletion of myeloid HIF-1α eliminated joint swelling and reduced synovial infiltration, pannus formation, and subsequent cartilage destruction. Loss of inflammatory capacity correlated with a defective metabolic activity without affecting development and differentiation of myeloid cells.
Hypoxia/HIF mediates recruitment of monocytes and lymphocytes
Migration of effector cells to the synovium is one of the clinical features of RA, as previously mentioned. Stromal cell derived factor 1 (SDF-1; or CXCL12) is a potent chemotactic and angiogenic factor that is expressed in RA synovial fibroblasts and contributes to the recruitment and accumulation of monocytes, T and B lymphocytes to the RA synovium.85–87 Underlining the relevance of hypoxia and HIF-1α in arthritis, Hitchon et al demonstrated the hypoxia dependent expression of HIF-1α and SDF-1 in RA synovium by immunohistochemistry. They observed the coexpression of SDF-1 and HIF-1α in consecutive sections and the increased SDF-1 messenger RNA levels in synovial fibroblasts.22 Recently, Ceradini et al demonstrated the HIF-1 and HIF-2 dependent SDF-1 expression in endothelial cells by promoter analysis.88
In addition to the chemoattractant molecule SDF-1, its receptor, chemokine receptor CXCR4, has been reported to be HIF-1α inducible, as has another migratory compound, the β2 integrin subunit CD18, which also has a role in the recruitment of myeloid cells to sites of inflammation.89,90 These findings suggest hypoxia/HIF induced accumulation and positioning of monocytes, T and B cells in the rheumatoid synovium. However, conclusive evidence for this process in RA is still lacking. Another factor leading to lymphocyte accumulation during hypoxia is the enhanced promotion of cell survival, as shown for T cells. Triggered by repeated antigen challenge through the T cell receptor complex (TCR/CD3), T cells undergo activation induced cell death during normoxia. During hypoxia, the induction of HIF-1 promotes T cell survival through increased expression of its target gene product adrenomedullin.24
HIF promotes arthritis by induction of cartilage destruction
One critical symptom in the pathogenesis of RA is the massive destruction of the articular cartilage. Cartilage degeneration is associated with high activity of so called matrix metalloproteinases (MMPs) (reviewed by Sato91). MMPs promote extracellular matrix remodelling essential for the sprouting of new capillaries during angiogenesis. MMP expression is mainly induced by the transcription factor Ets-1, which mediates tumour invasion and supports arthritis as one responsible factor involved in both aggressive pannus invasion and cartilage and bone destruction during arthritis.92 Ets-1 has been shown to be inducible by HIF-1.93 In addition, Ets-1 co-localises with HIF-1α in the inflammatory infiltrate and invasive pannus of the inflamed RA joint, as demonstrated in an AIA rat model.7 Thus, it can be suggested that HIF-1 promotes pannus invasion, cartilage destruction, and bone erosion through Ets-1.
HIF induces angiogenesis in arthritis (role of VEGF)
One outcome of HIF is the induction of angiogenesis, which has a pivotal role in the development and perpetuation of RA. Angiogenesis can be defined as a multistep process, which involves endothelial cell activation, increased blood vessel permeability, and local rearrangement of the basal membrane and extracellular matrix. Thus, endothelial cells migrate to, and proliferate at, the origin of angiogenic signals, influencing tissue remodelling and formation of new capillaries. Pro- and antiangiogenic mediators balance this process. In this context, HIF directly induces the expression of a broad range of pro-angiogenic factors like VEGF and its receptor Flt-1, Tie-2 and its receptor Flk-1, angiopoietins (fig 1), all of which are up regulated in the pathogenesis of RA.94,95
“Promotion of angiogenesis may be the most important role of HIF in the pathogenesis of RA”
Promotion of angiogenesis may be the most important role of HIF in the pathogenesis of RA. Inhibition of angiogenic processes in the inflamed joint in several RA models has been shown to reduce arthritis, which underlines the role of HIF, also known to be a key regulator of angiogenesis (reviewed by Koch96).
Inflammatory mediators can induce HIF
As previously mentioned, HIF can be induced under hypoxic but also normoxic conditions by several inflammatory factors, such as (a) ROS (for example, HO*, H2O2) and RNS (for example, nitric oxide), released during the respiratory burst by macrophages and neutrophils in inflammation97,98; (b) proinflammatory cytokines (IL1β or TNFα), which are abundant during inflammation induced joint disease15,18,22,99; and (c) pathogens such as bacterial lipopolysaccharide and hepatitis B virus X protein (see fig 5).100,101
By differential screening of a human synovial fibroblast cDNA library, HIF-1α was identified as a clone, which is up regulated by IL1. In addition, IL1 increases the binding of the heterodimer HIF-1 to the HIF consensus sequence.99 In cultured synovial fibroblast explants VEGF production was potently induced after treatment with transforming growth factor β, and to a lesser extent with IL1β and TNFα, which was further augmented by hypoxia.22 This may be one reason for the perpetuation of arthritis even under repeated cycles of hypoxia-angiogenesis-reperfusion.
In summary, HIF is expressed in the inflamed joint in synovial fibroblasts, macrophages, and CD3+ T cells. Its loss in macrophages and neutrophils reduces joint inflammation. HIF mediates cell migration in cancer; however, a direct link between HIF and cell migration in arthritis has been suggested but not proved. HIF can promote cartilage destruction by induction of ETS-1 and directly promotes angiogenesis through, for example, VEGF, its receptors, and the angiopoietin-Tie system.
THERAPEUTIC ASPECTS
A better understanding of the role of HIF-1α, and its induction and stabilisation/degradation processes involved in cellular functions of inflammatory reactions will suggest new targets and new therapeutic strategies that may overcome persistent and chronic inflammatory diseases like RA.
Commonly used drugs and clinical trials in RA
Today, drugs given in clinical practice and trials to treat RA are divided into several subgroups:
-
Biological response modifiers (anti-TNF drugs: etanercept, adalimumab, infliximab; selective IL1 blocker: anakinra; modulator of T cell stimulation: CTLA4Ig; B cell depletion agent: rituximab)
-
Glucocorticoids (for example, dexamethasone)
-
Disease modifying antirheumatic drugs (for example, methotrexate, ciclosporin; cyclo-oxygenase-2 (COX-2) inhibitors—for example, celecoxib
-
Non-steroidal anti-inflammatory drugs (for example, ibuprofen, indometacin, COX-2 inhibitors).
Given alone or in different combination they have systemic immunosuppressive and antiangiogenic effects, which are beneficial but not without severe side effects, such as osteoporosis, secondary infections, or enhanced risk of myocardial ischaemia. Thus, clinical administration always strikes a balance between the advantages and disadvantages of a given drug.
Direct targeting angiogenesis has been proposed using different types of antiangiogenic substances in in vitro studies and in vivo animal models of collagen and AIA (table 1).96 Indeed, many substances inhibiting induction of VEGF, direct action of VEGF or its receptor tyrosine kinase signalling are tested in clinical trials for cancer and for arthritis treatment.94 Anti -VEGF antibodies have been shown to delay the development of murine collagen induced arthritis (CIA) and to attenuate its severity when given before the onset of disease. After onset of disease, clinical and histological measures of arthritis improved.102 Another approach is the inhibition of the cytokine induced secretion of VEGF, which has been documented for transforming growth factor β, IL1, and TNFα in cultured synovial fibroblasts.22,103 Anti-TNFα treatment has been shown to reduce VEGF, vascularity, and the number of swollen joints, providing one possibility to suppress angiogenesis.102 Nevertheless, direct inhibition of HIF could lead to more efficient reduction of arthritis, targeting not only angiogenesis but also cell migration and cell homoeostasis.
Some angiogenesis inhibitors and their effect on HIF expression
HIF targeting agents: lessons from cancer treatment
Systemic targeting of HIF-1α always runs the risk of damaging physiologically hypoxic tissues—for example, lymph nodes, the thymus, or cartilage.104 Therefore, only local administration of anti-HIFα agents will be potentially beneficial.105 Nevertheless, some of the drugs mentioned above affect HIF, such as ciclosporin. Ciclosporin A inhibits hypoxia dependent gene transcription in a reporter gene assay and prevents the hypoxic accumulation of HIF-1α. It destabilises HIF-1α by promoting its hydroxylation.106 Similar effects were found in in vitro studies using the non-steroidal anti-inflammatory drugs, indometacin, ibuprofen, and a COX-2-specific inhibitor (NS-398), also suggesting a role of cyclo-oxygenases or their products in the activation of HIF.107–109 Major effects were achieved by directly targeting HIF with small molecules bearing cytotoxic and antiangiogenic effects. Substances like PX-478 and TX-402 as well as the HSP90 inhibitors 17-AAG have been shown to reduce HIF and HIF target gene transcription.110–113
A new vascular agent called YC-1 has already been shown to inhibit platelet aggregation and vascular contraction and to down regulate the expression of two HIF-1α target genes, EPO and VEGF in vitro. It also inhibits the expression of HIF-1α itself at the post-transcriptional level by an as yet undefined mechanism which may contribute to the down regulation of EPO and VEGF. The inhibition of HIF-1α activity in tumours of mice treated with YC-1 is associated with blocked angiogenesis and an inhibition of tumour growth. YC-1 has the potential to become the first antiangiogenic agent directly to target HIF-1α.114,115
The group of Claire E Lewis proposed targeting inflamed joints with therapeutic genes using motile cells. In view of the abundant HIF-1α expression in macrophages of the RA synovium, their aim is to generate transduced mobile macrophages with therapeutic genes—that is, antiangiogenic factors, under the control of a hypoxia inducible enhancer.7,21,116 Obviously, this approach will be restricted to local inflammation. Recently, the endogenous antiangiogenic protein endostatin has been shown to provide beneficial effects similar to those of YC-1 and reduce tumour growth. Because angiogenesis and vascularisation of cancer is essentially identical to that observed in RA, endostatin treatment potentially could be used for treatment of RA. Indeed, using a new model of RA, in which human RA tissue is grafted into severe combined immune deficiency mice, it was shown that human recombinant endostatin decreased the number of blood vessels and the number of inflammatory cells (macrophages and lymphocytes) and increased the number of apoptotic cells in RA synovia.117 Similar results were observed by Kurosaka et al in a CIA model in BALB/c mice, where systemic administration of endostatin inhibited the arthritis, particularly pannus formation and bone destruction.118 Moreover, Abdollahi et al treated human microvascular endothelium with endostatin and established a genome-wide expression pattern profile analysing HIF-1α, FIH, and several HIF target genes. Interestingly, all of these genes were down regulated. This, together with the in vivo results from the mice with CIA, suggests that endostatin may be an endogenous direct inhibitor of HIF and HIF target gene expression.119
CONCLUSION AND PERSPECTIVES
Reviewing the structure and functional role of HIF-1α as a key player in cell homoeostasis and cell function, we aimed at underlining the important role of this oxygen sensitive transcription factor in immune cell function for pathogenesis, progression, and persistence of arthritis. Elucidating the effects of hypoxia on HIFα activity could provide new strategies in arthritis treatment using HIFα and/or its inductive pathways as possible targets. However, before efficient anti-HIF drugs or therapeutic gene targeting scenarios will be applicable for clinical trials, several aspects of HIF induction, signalling, and effects have to be elucidated, especially for the pathological circumstances in arthritis. Does up regulation of HIF-1α during RA result from ROS induced mutations in genes or promoter regions, as observed for p53? Are HIF-2 and HIF-3 expressed in a similar way to HIF-1 in the inflamed joint? And if so, is there a correlative link with RA? Furthermore, the role of the negative regulators of HIF, pVHL, FIH-1, and hIPAS has not yet been fully investigated, and their role in arthritis, especially, remains a matter for research.
Acknowledgments
Supported by research grant 01GS0110/ 01GS0160/ 01GS0413 from the Federal Ministry of Education and Research, Germany.