Article Text

Abstract
In this viewpoint, we summarise three different lines of evidence suggesting that current biological therapies directed at different molecules or cells have similar efficacy in rheumatoid arthritis and target similar populations of patients; therefore, distinct biological effects of targeted therapies may not account for differences in response. Moreover, currently available individual biomarkers or multiple biomarker sets do not provide information beyond that conveyed by clinical disease activity. Smart and novel research designs will have to be developed to find pertinent biomarkers. Until then, the focus of clinicians may have to solely rest on clinical disease activity assessment and targeting remission or low disease activity rapidly.
- Rheumatoid Arthritis
- Cytokines
- DMARDs (biologic)
- Outcomes research
- TNF-alpha
Statistics from Altmetric.com
Synovitis is the harmful foundation of rheumatoid arthritis (RA). It causes swelling, pain and stiffness and is directly responsible for the cartilage and bone destruction so characteristic of this disease.1 Clinical symptoms and joint damage cause the functional impairment that interferes with RA patients’ quality of life and working capacity. Thankfully, patients and rheumatologists have a variety of disease modifying drugs (DMARDs) to choose from today, both synthetic small chemicals as well as biologic;2 and aside from the latter, more targeted therapies, including small drugs, are on the horizon.3
With the efficacy of all these agents and with focusing treatment to the target of low disease activity or remission in mind,4 RA patients have excellent prospects already now and even more so in the future. Still, many patients have active disease and suffer from increasing disability. Beliefs that this is the consequence of bad access to optimal care, such as lack of reimbursement of biological agents, may have some grounds, but may not necessarily be the major driver of the problem. Rather, lack of adherence to treating RA to the above mentioned targets might be the culprit behind the scene. Studies like TICORA, BeSt and more recently CAMERA 2 are the best witnesses of this conclusion.5–7
Some observations support this inference: in clinical trials of patients with an insufficient response to methotrexate (MTX), the DMARD history reveals that despite long disease duration only few DMARDs had been employed (table 1).8–13 Of course, while no details on the disease history were presented and the disease could have been under good control for longer periods of time, we know for some of the trials that patients must have had quite active disease over time, given the relatively large extent of their joint damage at baseline (table 1). Thus, thousands of patients worldwide enter clinical trials who have active and highly destructive disease and, for whatever reason, apparently have received only very few DMARDs in the course of their RA.
Presentation of some baseline data of randomly selected clinical trials and calculation of the average duration that each disease modifying antirheumatic drug (DMARD) was employed
American College of Rheumatology (ACR)50 and ACR70 response rates and frequencies of serious adverse and serious infectious events in trials on combinations of biological agents14–16
On the other hand, even when rheumatologists have all approved drugs available, they still have no or little clues to decide which one to choose. All biological agents offer about the same frequency of good outcomes, if—for example—one takes an American College of Rheumatology (ACR) 50% or 70% response rate into consideration.17 Therefore, many clinicians believe that better markers—ideally biomarkers—would allow more appropriate selection of the right medicine. Indeed, personalised medicine is today's major general thrive in RA and other diseases.18–21 Are biomarkers, which by the way we have failed finding definitively hitherto, truly going to be the solution of the dilemma?
To address this issue, let us consider three aspects related to the major enticing question: Do all patients have the same chance of response with all specific drugs? In other words, is there one pool of responding individuals, are the responder pools distinct or do they overlap?
-
In tumour necrosis factor (TNF)-blocker failures, any biological mode of action will work to a similar extent, including TNF-inhibitors. If the chance of success on a particular drug was distinct for different patients, then those who failed one type of a targeted therapy should respond better to another targeted principle. In other words: patients who have failed a TNF-inhibitor should be responding better to other principles than to a TNF-blocker. The reality is different: when looking at comparable populations of patients continuing to receive MTX, then the overall ACR50 responses at 24 weeks are very similar irrespective of the targeted treatment used (figure 1A). Evidently, this is just a comparison of published data and anything but a conclusive head-to-head trial, but direct comparative trials25 have reaffirmed that an ‘eyeballing approach’ may be informative in the absence of direct comparisons. While some observational studies have shown that after failure of one or at least two anti-TNF agents the choice of a biological agent with a different mechanism of action could lead to better clinical results,26 ,27 there are a number of confounding factors inherent to such analyses. First, observational studies and registries do not evaluate randomised patient populations, and registries were primarily developed to understand long-term safety aspects in a real world; second, in a study from a registry, for example, which suggested that rituximab may be more effective than another TNF-blocker in patients who failed initial TNF-inhibitor therapy, almost 40% of the patients receiving a TNF-blocker did not have concomitant DMARD therapy;26 however, it is well established that anti-TNF plus MTX combination therapy conveys significantly higher efficacy when compared with monotherapy, and this is not likely to be correctable for statistically28 ,29 third, systematic literature reviews and pairwise meta-analyses of clinical trials did not show differences in this regard.30 ,31 Therefore, currently no firm evidence exists that more patients who previously failed a TNF-blocker respond to therapies directed at another target than TNF compared with a TNF-inhibitor. Thus, there is no evidence for different pools of patients.
-
Response to most biological modes of action decreases with the number of failed prior TNF-blocker courses. Likewise, if there were such different patient pools, efficacy of a new treatment principle should not decrease with the use of increasing numbers of TNF-inhibitors prior to starting that new principle. Alas, with one exception, currently available data suggest that responsiveness decreases with increasing numbers of prior TNF-blocker therapies. Such decrease has been observed for rituximab32 and abatacept,33 and patients with three prior TNF-blocker therapies also responded less to golimumab than patients who received one or two prior TNF-inhibitors.34 While the lower response rates with increasing numbers of failed therapies might be the consequence of an increase in general refractoriness to any therapy with increasing numbers of treatment cycles, this has not yet been unequivocally established and the observation that with tocilizumab the response rates did not decrease with more prior TNF-inhibitor therapies22 speaks against this assumption, although these data come from a single study and might also reflect a play of chance. A further recent publication again suggested that rituximab may be most effective in patients who failed no more than one TNF-blocker:35 if the difference in mode of action is a major reason for therapeutic success, why should this not be even better when more than one anti-TNF failed? Therefore, again, evidence that patients who are responsive to other targeted therapies are generally drawn from a different subset than patients responding to a second or third TNF-inhibitor is currently lacking, at least for costimulation blockade and B cell depletion.
-
Combinations of biological agents do not recruit additional responders. The probably most compelling argument rests with the third item: is a combination of two different targeted biological therapies more efficacious than just one of them? If patients preferentially respond to one therapy but not to another one, thus belonging to different responder pools, then combinations of biologics should increase efficacy. However, this has consistently not been the case according to three randomised controlled trials. The first one compared monotherapy of etanercept head-to-head to full or half doses of etanercept combined with anakinra, an interleukin (IL)-1-inhibitor.14 Not only was there no added benefit of the combination, but etanercept monotherapy consistently had numerically higher response rates than any of the combination groups (table 2). This lack of added efficacy was not due to insufficient IL-1 blockade or an overriding effect of TNF inhibition, since the dramatic increase of serious adverse events including serious infections indicated an added biological effect of the combination. In a more recent trial, patients with active disease despite etanercept or adalimumab therapy in combination with MTX were randomised to receive rituximab or placebo. No patient in either group achieved an ACR70 response, and ACR50 responses were quite low in both treatment arms (table 2); compared with a previous study on rituximab in patients who have had prior TNF-blocking treatment,23 the response rates were numerically much lower. The rates of serious adverse events were low in this small study, but numerically higher in the combination therapy group.
A third clinical trial assessed combination of a TNF-inhibitor with T cell costimulation blockade.36 The differences in ACR response rates did not reach statistical significance; importantly, while in the double blind phase abatacept was used only at 2 mg/kg, during long-term extension the dose was 10 mg/kg and there was still no significant clinical difference compared with etanercept plus placebo. However, serious adverse events, including serious infections, were much more frequent in the combination than the monotherapy group.
These data reveal that combinations of TNF-inhibitors with T cell, B cell or cytokine directed therapy do not convey added benefit but increase the risk of side effects. This increase in serious adverse and infectious events, revealing indeed an added biological effect of all these combination therapies, in conjunction with a lack of significant clinical benefit is so sobering.

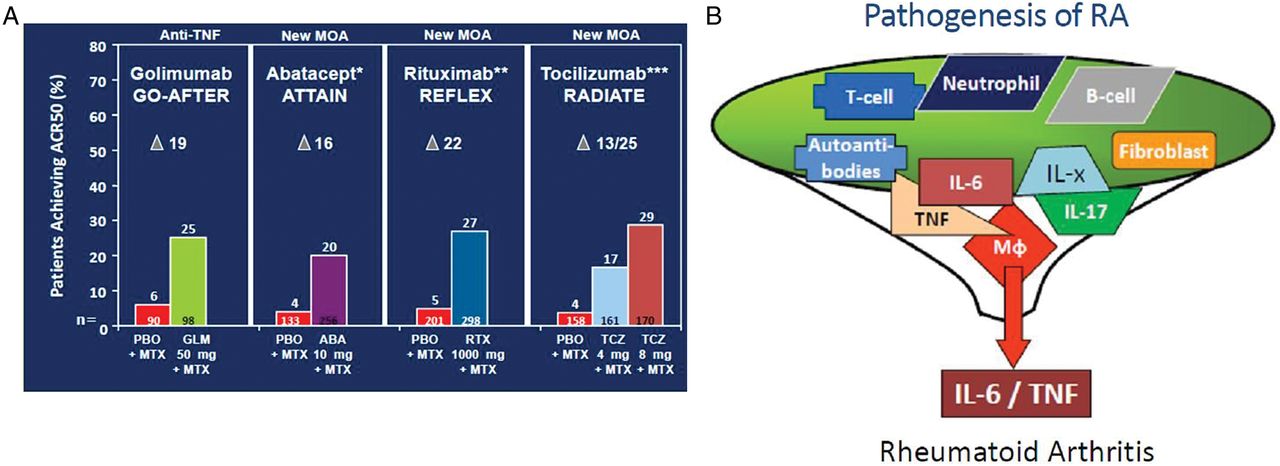{kind=link}
Different biological agents have similar efficacy and may affect a common final pathway. A: ACR50 response rates at 24 weeks as published for trials on golimumab, abatacept, rituximab and tocilizumab in combination with methotrexate (MTX).11 ,22–24 Of note, the primary endpoint of the golimumab trial was at 14 weeks and comprised patients with and without synthetic disease modifying antirheumatic drug therapy; the data shown here relate to a secondary endpoint at 6 months of patients treated with golimumab plus MTX11 to ensure comparability with the other trials. The numbers shown at the bottom of each column show the numbers of patients studied. The Greek delta denotes the difference between active therapy and placebo B: Schematic presentation of several individual pathogenetic molecules or cells funnelling into a single final pathway leading to rheumatoid arthritis. ACR, American College of Rheumatology; IL-x, other cytokines.
Taken together, the answers to the three questions are quite consistent and suggest that patients who do well upon treatment with any of the currently available biological agents are mostly drawn from the same pool of responders. Why should this be the case for therapies whose targets differ so much? Because all these pathogenetic principles may funnel into a common final pathway which is interfered with whether the target is further upstream or further downstream, as long as it is pathogenetically important (figure 1B). Of note, rituximab is somewhat more efficacious in rheumatoid factor positive than negative RA;37 but first, this viewpoint primarily relates to the about 80% or so seropositive RA patients, who may have a different disease than seronegative ones, and second, a recent meta-analysis of rituximab revealed that the difference in efficacy between seropositive and seronegative patients may be modest;37 moreover, rituximab has shown some efficacy even in ankylosing spondylitis and psoriatic arthritis, two seronegative diseases.38 ,39
On all these grounds, distinct biological effects of targeted therapies may not account for differences in response and, as a consequence of these deliberations, the search for biomarkers allowing one to target specific populations at risk might be less revealing than we would hope. Moreover, we have to bear in mind that the best predictor of reaching remission or low disease activity is not an individual or a group of biomarkers, but the reduction of disease activity by composite measures itself,40 and the higher the baseline disease activity, the lower the chances of a good outcome. This latter statement relates to all types of therapies, again suggesting common grounds for responders. And while the search for markers needs to be continued in a more innovative way than hitherto, we just have to accept that currently, rather than biological markers, clinical markers appear to be most predictive of outcome and response to therapy. Since the major task of RA treatment is to reduce disease activity, ideally reaching a state of remission or low disease activity, we need to focus mainly on this task and not be too disappointed if we fail to find biomarkers.
In light of the frequently observed prolonged use of individual DMARD therapies over an average of about 3 years despite apparent insufficient efficacy, as discussed above, we should not focus too much on biomarkers currently known to be at least partly associated with disease activity or treatment response (acute phase reactants, autoantibodies, cytokine profiles41) or wait for new biomarkers to be detected. As of now, even elaborated biomarker assessments have not been shown to expand the information provided by clinical measures of disease activity42 nor—maybe with few exceptions41—to be highly predictive of therapeutic response, and have partly been afflicted with lack of sensitivity and interference with factors present in serum of RA patients.43 Thus, we should currently rather focus our attention on just reducing disease activity in a treat-to-target approach, which includes adapting or changing therapy every 3–6 months if improvement (within 3 months) or the target of at least low disease activity (by 6 months) is not attained.
These considerations are not at all meant to be discouraging regarding the search for markers that may be helpful to develop a personalised medicine approach in RA: they just reflect the current evidence base. And they imply that smart and specifically focused clinical trial designs and research projects will be needed to detect biomarkers which allow one to differentiate responsiveness to one but not another biological agent. Until such markers are detected, abating disease activity, stopping progression of joint damage and maximising physical function and quality of life should come into focus more strongly than currently frequently realised, irrespective of any type of markers or too much emphasis on modes of action of certain drugs. Some markers will come and others will go—fighting active disease will remain at the heart of our pursuits.
References
Footnotes
-
Competing interests JS has received grant support and/or provided expert advice and/or presentations for Abbott, Astra-Zeneca, BMS, Celgene, Glaxo, Janssen, Lilly, Medimmune, MSD, Novartis-Sandoz, Novo-Nordisk, Pfizer, Roche, Teva and UCB. DA has provided expert advice and presentations for Abbott, Grunenthal, MSD, Medac, Pfizer, Roche and UCB.
-
Provenance and peer review Not commissioned; externally peer reviewed.