Article Text

Abstract
Treatment of pain in rheumatoid arthritis must take into account the gastrointestinal and cardiovascular risk of individual patients. Adequate results are not yet available, and until they are, treatment recommendations must take into account, not only the more favourable gastrointestinal risk profile of selective COX-2 inhibitors, but also the potential atherothrombotic risk of any NSAID or selective COX-2 inhibitor treatment.
- APC, Adenoma Prevention with Celecoxib
- APPROVe, Adenomatous Polyp Prevention on Vioxx
- ASA, acetylsalicylic acid
- bid, twice a day
- CABG, Coronary Artery Bypass Graft
- CI, confidence interval
- CLASS, Celecoxib Long term Arthritis Safety Study
- COX-2, cyclo-oxygenase-2
- CV, cardiovascular
- GI, gastrointestinal
- IV, intravenous
- MI, myocardial infarction
- NSAIDs, non-steroidal anti-inflammatory drugs
- PGI2, prostaglandin I2
- PPI, proton pump inhibitor
- PreSAP, Prevention of Spontaneous Adenoma Polyps
- RA, rheumatoid arthritis
- tid, three times a day
- TARGET, Therapeutic Arthritis Research and Gastrointestinal Event Trial
- TXA2, thromboxane A2
- VIGOR, Vioxx Gastrointestinal Outcomes Research
- non-steroidal anti-inflammatory drugs
- atherothrombotic risk
- COX-2 inhibitors
Statistics from Altmetric.com
- APC, Adenoma Prevention with Celecoxib
- APPROVe, Adenomatous Polyp Prevention on Vioxx
- ASA, acetylsalicylic acid
- bid, twice a day
- CABG, Coronary Artery Bypass Graft
- CI, confidence interval
- CLASS, Celecoxib Long term Arthritis Safety Study
- COX-2, cyclo-oxygenase-2
- CV, cardiovascular
- GI, gastrointestinal
- IV, intravenous
- MI, myocardial infarction
- NSAIDs, non-steroidal anti-inflammatory drugs
- PGI2, prostaglandin I2
- PPI, proton pump inhibitor
- PreSAP, Prevention of Spontaneous Adenoma Polyps
- RA, rheumatoid arthritis
- tid, three times a day
- TARGET, Therapeutic Arthritis Research and Gastrointestinal Event Trial
- TXA2, thromboxane A2
- VIGOR, Vioxx Gastrointestinal Outcomes Research
Vascular atherosclerosis is an inflammatory disorder associated with characteristic lesions of the vessel wall, which induces cellular interactions that do not differ fundamentally from those of other chronic inflammatory fibroproliferative diseases.1 Inflammatory processes mediated by cyclo-oxygenase-2 (COX-2) are inhibited by traditional non-steroidal anti-inflammatory drugs (NSAIDs) or COX-2 selective inhibitors. This can halt the atherogenesis in its early stages.2,3
Prostacyclin (prostaglandin I2 (PGI2)) is a vasodilator that inhibits platelet function. However, inhibition of PGI2 synthesis does not lead to spontaneous thrombosis.2 In endothelial cells PGI2 synthesis is mediated by COX-2, which in turn is haemodynamically induced or activated by oestrogen.4 PGI2 modulates platelet–vascular interactions and specifically limits the response to thromboxane A2 (TXA2). Selective COX-2 inhibitors inhibit PGI2 but not TXA2.5 Selective COX-2 inhibitors reduce PGI2 dependent atheroprotective processes such as platelet aggregation inhibition and vasodilatation and decrease the proliferation and contraction of smooth muscle cells. COX-2 inhibitors promote interactions between neutrophils and platelets and the vessel wall thus contributing to atherogenesis.4,6 In premenopausal women chronic treatment of patients with selective inhibitors of COX-2 could undermine protection from cardiovascular disease.4,5
Unlike selective COX-2 inhibitors, NSAIDs reversibly inhibit the production of TXA2 in platelets. However, the resulting decrease in platelet aggregation does not generally persist beyond the overall dosing interval.7 Moreover, the correlation between NSAID-induced inhibition of TXA2 production and platelet functions is not linear. The imbalance between PGI2 and TXA2, which is said to be the reason for the atherogenic potential of selective COX-2 inhibitors, is also likely to exist in large segments of the dosing intervals during NSAID treatment.8 A possible exception is naproxen (500 mg twice daily (bid)), which—at least under study conditions—can attain stable and sufficiently high plasma concentrations to compensate for the PGI2/TXA2 imbalance.9 With most NSAIDs, therefore, an increase in the thrombogenic risk must be expected. The results of large outcome studies and of more recent intervention studies, have to be seen in this light, and treatment recommendations should be modified accordingly.
ROFECOXIB
In the Vioxx Gastrointestinal Outcomes Research (VIGOR) study, the gastrointestinal (GI) superiority of rofecoxib (50 mg daily) over naproxen (500 mg bid) was demonstrated in 8076 patients with rheumatoid arthritis (RA) treated for a median period of 9 months. Rates of complicated confirmed events (perforation, obstruction, and severe upper GI bleeding) were 0.6 and 1.4 per 100 patient-years, respectively (p = 0.005). In that study the incidence of cardiovascular (CV) thrombotic events doubled during treatment with rofecoxib. Myocardial infarction (MI) occurred more frequently with rofecoxib than with naproxen (0.4% v 0.1%; 95% confidence interval (CI) 0.1 to 0.6). There was no correlation between MI and hypertension, and CV mortality and cerebrovascular ischaemia occurred in 0.2% of patients in both groups.10 The difference in the incidence of MI was a secondary outcome in the study and may have been a chance finding. Biomedical models suggest two other seemingly contradictory yet plausible hypotheses for a possible atherogenic effect with rofecoxib and a cardioprotective effect with naproxen that is comparable to the effect of aspirin (acetylsalicylic acid (ASA)).9–,11
A retrospective analysis found that 4% of study participants had a history of CV disorders and were included in the study contrary to the protocol. In accordance with the protocol, these patients were not treated with ASA. Thirty eight per cent of the MIs occurred in this high risk group.10 Rofecoxib may have unmasked the thrombogenic potential in these high risk patients and may even have potentiated the thrombotic potential compared with celecoxib in the CLASS study, where ASA was permitted.8 In the remaining patients, the incidence of MI was not significantly different: 0.2% with rofecoxib and 0.1% with naproxen.10
The Adenomatous Polyp Prevention on Vioxx (APPROVe) study included almost 2600 patients and started 9 months after the approval of rofecoxib in America and 1 month before the results of the VIGOR study became known.11,12 The CV risk increased, for the first time, 18 months after the start of treatment with 25 mg rofecoxib daily. After a further 18 months of treatment with rofecoxib, the difference attained significance (p = 0.008). The incidence of severe thromboembolic events was 1.92 times higher in those treated with rofecoxib than in the placebo group.12 In September 2004 the study was stopped prematurely and the manufacturer withdrew the medicine from the market.
CELECOXIB
The outcome of the Celecoxib Long term Arthritis Safety Study (CLASS) was similar to VIGOR performed in 8059 patients. The annual incidence of upper GI ulcer complications combined with symptomatic ulcers for celecoxib (400 mg bid) v NSAIDs (diclofenac 75 mg bid and ibuprofen 800 mg three times a day (tid)) was 2.08% v 3.54% (p = 0.02). However, CLASS included an insufficient number of participants to achieve the primary target criterion—namely, a significant reduction in the incidence of upper GI ulcer complications between celecoxib alone versus two traditional NSAIDs combined with permitted ASA treatment. In the 6 month treatment period, the incidence of MI CV events in the celecoxib group (0.9%) did not differ from that in the NSAID group (1.0%). Among those patients not treated with ASA, CV events occurred with equal frequency (0.5% with celecoxib and 0.4% with NSAIDs).13 Any existing atherogenic potential of celecoxib may have been masked in the 21% of participants using ASA.8
“In the CLASS study the potential of celecoxib may have been masked by patients using aspirin”
The Adenoma Prevention with Celecoxib (APC) study was carried out by the National Cancer Institute in 2035 patients and was prematurely stopped by the National Institutes of Health after an average treatment period of 33 months. The study was stopped because the incidence of CV events (CV death, MI, stroke) showed a dose dependent 2.3-fold and 3.4-fold increase during celecoxib treatment in the 200 mg bid and 400 mg bid dose groups, respectively, compared with the placebo group.14 Additionally, in the APPROVe study, CV events also occurred dose dependently and with a similar frequency in 1–2% of patients treated with celecoxib. A second placebo controlled study (Prevention of Spontaneous Adenoma Polyps (PreSAP)) with a comparable study design did not show any increased CV risk with celecoxib at a dose of 400 mg daily after a similar mean treatment period.15,16
The placebo controlled, three arm Alzheimer’s Disease Anti-Inflammatory Prevention Trial (ADAPT), which was sponsored by the National Institute of Aging, was also stopped by the National Institutes of Health as a precautionary measure in December 2004. The study included about 2400 volunteer subjects (mean age about 70 years) who were treated with naproxen (220 mg bid) or celecoxib (200 mg bid). In this study it was the naproxen group that showed a significant increase in CV risk compared with placebo. The celecoxib group showed no abnormal findings in this respect.17
VALDECOXIB
In the placebo controlled Coronary Artery Bypass Graft (CABG)-1 study,18 patients received parecoxib (40 mg intravenously (IV) for ⩾3 days), followed by valdecoxib (40 mg bid orally for 14 days), immediately after their coronary bypass operation. This treatment regimen was used in a modified form in a second study (CABG-2).19 A loading dose of 40 mg parecoxib by IV injection was followed by a period of at least 3 days in which 20 mg parecoxib was administered by IV injection every 12 hours, which was followed in turn by 10 days of oral treatment with valdecoxib (20 mg bid). Every phase of the study was placebo controlled. The incidence of severe CV events in the parecoxib/valdecoxib group (2.2% and 2%, respectively) was significantly higher than in the placebo/placebo group (0.0% and 0.5%, respectively). After this study, a warning about CV risk was included in the prescribing information.20
In a third study, a controlled treatment schedule was employed following general surgical interventions that was comparable to the regimen defined in the CABG-2 protocol. No differences in the incidence of CV events between placebo/placebo and parecoxib/valdecoxib were found.21 Moreover, no sufficiently conclusive CV safety data are available for long term treatment with lower valdecoxib doses in patients with relatively minor CV risk.21,22
The lack of adequate data on the CV safety of long term use of valdecoxib, and the increased risk of adverse CV events in CABG trials, together with the increased risk of rare but serious unpredictable skin reactions associated with valdecoxib, already described in its label, seems to demonstrate a lack of any advantages for valdecoxib compared with other NSAIDs. In April 2005 the manufacturer agreed to suspend the use of valdecoxib in Europe and the United States as an interim measure pending finalisation of the assessment of COX-2 inhibitors.23,24
LUMIRACOXIB
The Therapeutic Arthritis Research and Gastrointestinal Event Trial (TARGET) included 18 325 patients and compared lumiracoxib (400 mg daily) with naproxen (500 mg bid) and ibuprofen (800 mg tid)25,26; 24% of patients received concomitant ASA. In patients treated with lumiracoxib, the incidence of upper GI ulcer complications was two thirds lower than in the patients treated with an NSAID, this difference being significant. In the subgroup of patients treated with concomitant ASA, no significant difference was found. This is not surprising as the number of cases was too small to settle this question.26
The CV end point (non-fatal and silent MI, stroke, or CV death27) was met by about the same number of patients in the lumiracoxib group as in the NSAID group (0.65% v 0.55%; p = 0.5074). Patients who did not receive cardioprotective treatment with ASA showed no statistically significant differences in CV events. In the naproxen substudy, a tendency towards increased MI was seen with lumiracoxib (0.21% v 0.38%; p = 0.1471) compared with the ibuprofen substudy, in which a lower incidence of MI was seen with lumiracoxib (0.16% v 0.11%; p = 0.4833).25
NAPROXEN AND OTHER NSAIDS
In the naproxen group of the VIGOR study, the incidence of CV side effects was significantly lower than in the rofecoxib group. The aspirin-like cardioprotective effects of naproxen were given as a possible explanation for this.10 Retrospective analyses and follow up studies supported this view.10,28–,32 An unequivocal confirmation of the atherothromboprotective activity of naproxen has yet to be demonstrated in clinical studies.
The outcome of the Alzheimer prevention study (ADAPT), which was announced at the end of 2004, showed a higher incidence of CV and cerebrovascular events with naproxen (220 mg bid) than with placebo over a period of 3 years,17 which is not surprising in view of the incomplete COX-1 inhibition and complete COX-2 inhibition. With regular ingestion, high doses of naproxen (500 mg bid) competitively and reversibly inhibit platelet COX-1 activity and TXA2 biosynthesis beyond the 12 hour dosing interval in a manner similar (in terms of its completeness) to the irreversible COX-1 binding achieved with low dose ASA. However, unlike ASA, the inhibitory effect rapidly subsides after the last naproxen dose.9 In practice, the cardioprotective effect cannot be ensured with low daily doses and irregular use of naproxen. In contrast with ASA, naproxen also inhibits COX-2 dependent PGI2 synthesis9 and thus weakens any atheroprotective potential.
What has been said for naproxen probably also applies in principle to other NSAIDs. Clinical studies to assess atherogenic potential have not been carried out with traditional NSAIDs and individual safety profiles need to be established for the various NSAIDs based on clinical studies.
“With most so-called ‘tried and tested’ NSAIDs an increase in the thrombogenic risk must be expected”
These studies must take into account pharmacokinetic properties (for example, half life, bioavailability) and molecular differences of NSAIDs. Non-enzymatic mechanisms contribute to the differences in atherogenic potential of some NSAIDs.32 For example, experiments show that in contrast with other selective COX-2 inhibitors (celecoxib, valdecoxib, meloxicam) or non-selective NSAIDs (ibuprofen, naproxen, diclofenac), sulfone COX-2 inhibitors (rofecoxib, etoricoxib) exert a pro-oxidative influence on low density lipoprotein oxidation, which promotes the pathogenesis of atherosclerosis.33 The clinical relevance of this, however, is not clear.
The need to assess the CV risk of an NSAID is not confined to the active treatment phase. The risk of a primary MI appears to be increased for several weeks after the withdrawal of NSAID treatment, especially if the treatment was long term and another systemic inflammatory disease is present at the same time. The cause is assumed to be a vascular rebound effect.34 The activation of platelet activation following the absence of COX-1 inhibition and TXA2 synthesis, as well as the flaring up of inflammatory processes in the coronary vessel wall with subsequent plaque instability, are possible reasons for the increase in the incidence of acute MI.
ASPIRIN
Aspirin (acetylsalicylic acid or ASA) acetylates a single serine residue in the COX-1 (Ser529) and the COX-2 channel (Ser516) and thereby permanently inactivates the enzyme.35 The resulting longlasting inhibition of TXA2 synthesis in anuclear platelets is the basis for the antithrombotic cardioprotective effect of low doses of ASA.9 Daily aspirin doses of 75–325 mg are regarded as suitable for inhibiting platelet aggregation as a means of cardioprophylaxis in patients at risk (acute MI, a history of MI, a history of stroke or transient ischaemic attacks or other relevant disorders or events such as unstable angina, vascular surgery, angioplasty, atrial fibrillation, heart defects, peripheral vascular disease, etc).27 When doses higher than 100 mg/day are used, both ASA and naproxen also inhibit COX-2 dependent PGI2 synthesis.27,36 The expected simultaneous suppression of TXA2 and PGI2 may reduce the cardioprotective effect of low dose treatment. The concomitant administration of ibuprofen, but not rofecoxib or diclofenac, antagonises the irreversible platelet inhibition induced by ASA. Treatment with ibuprofen in patients with increased CV risk may limit the cardioprotective effects of ASA.7
“The cardiovascular protective effects of aspirin may be limited by concurrent use of ibuprofen”
It is difficult to interpret the results of a recent placebo controlled study in which the use of ASA at doses of 81 mg and 325 mg was investigated over a 3 year period for the prevention of colorectal adenoma.37 In the 749 subjects treated with ASA, seven MIs and seven strokes (of which one was a haemorrhagic insult) occurred compared with only one MI among the 372 patients in the placebo group.37 When the published data are analysed according to the criteria of the Antiplatelet Trialists’ Collaboration (APTC), the difference is significant (p = 0.006, 95% CI 1.3 to 78).27 Coronary revascularisation was performed equally frequently in the ASA group (eight cases in 749 patients) and in the placebo group (four cases in 372 patients). This surprising result may be a chance finding. However, it may also be suggestive of an increased CV risk in at least some of the study participants treated with ASA.
For CV secondary prophylaxis, low dose ASA is prescribed, where appropriate, in addition to selective COX-2 inhibitors. Concomitant selective COX-2 inhibition causes the rate of gastroduodenal ulcers to rise close to that of a dual COX-1/COX-2 inhibitor alone.38 In the CLASS outcomes study, reductions in ulcer complications were not significant in those taking aspirin (0.79, p = 0.4876).13 In another study, celecoxib together with ASA (325 mg/day) induced significantly more ulcers at 1 week than ASA alone (18.7% v 7.6%), but significantly fewer ulcers than the non-selective NSAID naproxen plus ASA (18.7% v 27.3%).39 In the TARGET study, the GI advantage of lumiracoxib in the group treated with ASA was only discernible as a trend.26 The use of enteric coated rather than plain ASA does not decrease the risk of GI bleeding.40,41
LOWER GI TRACT
Serious lower GI events occurred at a rate of 0.9% per year in patients with RA taking the non-selective NSAID naproxen, accounting for nearly 40% of the serious GI events that developed in these patients. Serious lower GI events were 54% lower with the use of the selective COX-2 inhibitor rofecoxib.42 A clinically meaningful decrease in haemoglobin (>20 g/l) or packed cell volume (>10%) level was seen in significantly more patients taking ibuprofen (5.4%) than in those taking placebo, ASA, or ASA plus rofecoxib (0.8%–1.6%).38 In capsule endoscopic studies, celecoxib leads to a significant reduction in lower bowel lesions compared with the combination of naproxen with a proton pump inhibitor.43
ASSESSMENT AND RECOMMENDATIONS
The seemingly contradictory results of numerous studies on the atherogenic potential of various NSAIDs and selective COX-2 inhibitors may be, at least in part, explained by epidemiological differences in the study groups, the primary indications for treatment, differences in the length of the studies, and other factors dependent on the study design. Differences in the CV safety of NSAIDs should be studied prospectively using direct comparisons between NSAIDs, where possible, in three arm studies including placebo. These requirements are met, at least partly, by the outcomes studies performed to date with COX-2 selective inhibitors—namely, VIGOR,10 CLASS,13 and TARGET.25,26 The results of APPROVe,12 APC,14 PreSAP,15,16 ADAPT,17 CABG-1,18 and CABG-219 allow indirect comparisons of the agents to be made owing to the placebo arms (see table 1⇓). Contradictory results may arise from differing patient groups, which may also differ substantially from the licensed indications of the agents. Some non-CV primary study results, which have yet to be published, would possibly facilitate the benefit–risk evaluation.
Summary of findings in relevant studies discussed
Despite the inconsistency of their conclusions on CV safety, the listed studies have, nevertheless, provided crucial data for changes in the licences for these medicines. They have had an influence on pending approval procedures, treatment recommendations, and practical treatment decisions, which in addition to GI safety issues, now increasingly have to take CV aspects into account. The results of VIGOR and APPROVe may have been only the tip of the iceberg, concealing comparable CV side effect profiles of the so-called “tried and tested” NSAIDs. No studies are available which have been able to shed light on these questions, and none are likely to be performed because of the high costs involved. Precautionary notes have been included in the prescribing information, and CV signs such as oedema or hypertension are regularly listed as side effects in the prescribing information of traditional NSAIDs.
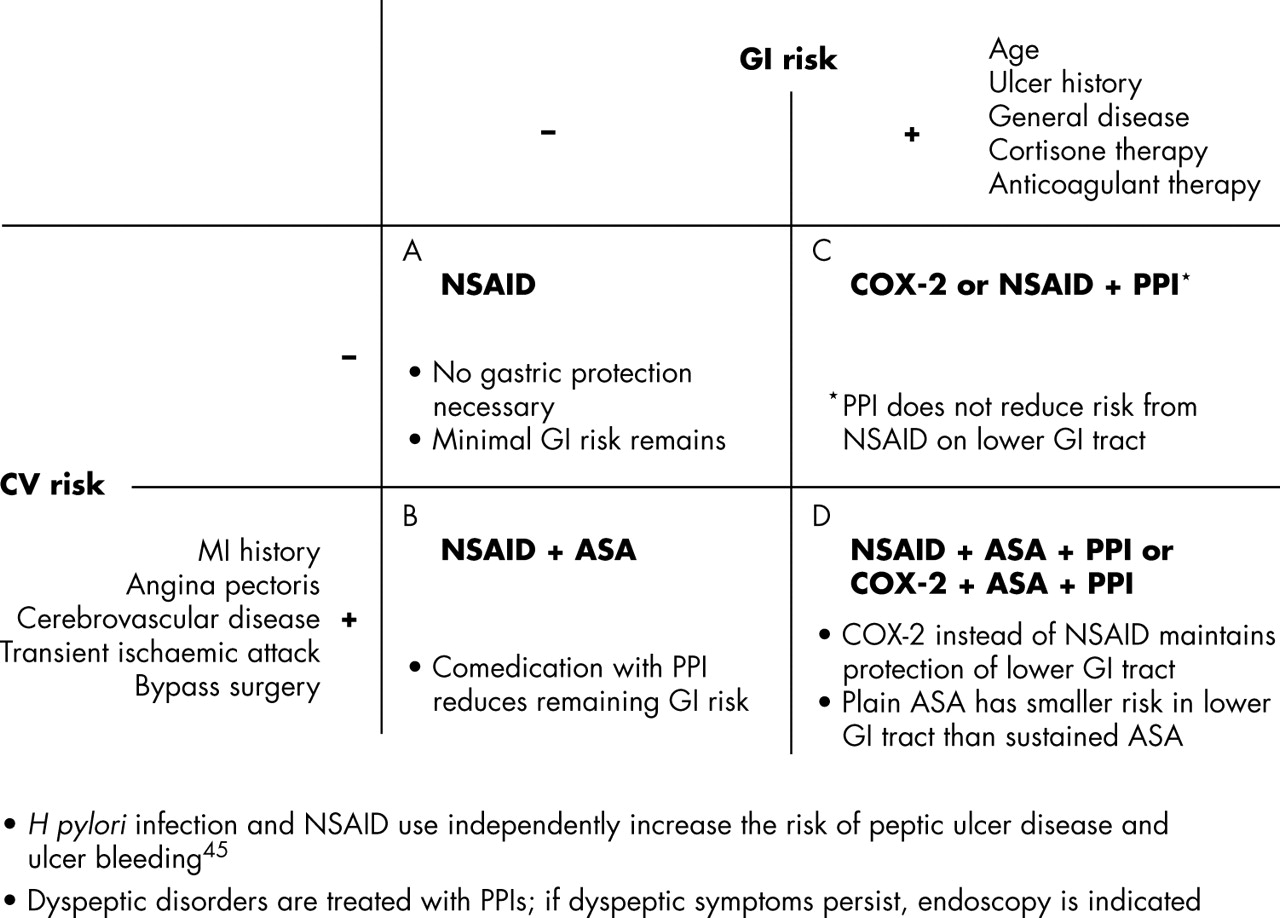
The new results from these studies influence recommendations for prescribing behaviour (see fig 1⇓). There is a real need to avoid making hasty conclusions about the safety of traditional NSAIDs. In the course of any NSAID treatment, the individual indication must be regularly reviewed. Unnecessary courses of treatment must be avoided, and alternatives with minimal side effects and adequate efficacy must be used. Selective COX-2 inhibitors remain a sensible choice for patients with low CV risk who have experienced severe GI events, especially during treatment with NSAIDs. Based on the data currently available, selective COX-2 inhibitors should not, as far as possible, be used in patients with CV disorders or increased CV risks.6 For the protection of patients requiring treatment, precautionary measures should be extended to include all NSAIDs and selective COX-2 inhibitors in what is a rapidly changing field, both scientifically and clinically. For this reason, “tried and tested” substances should be re-examined for their atherogenic potential. However, abrupt withdrawal of NSAIDs should be avoided because of possible vascular rebound effects in patients with systemic inflammatory disorders.34
{kind=link}
Treatment of RA inflammatory pain taking into account the individual GI and CV risk. (A) Without GI risk, traditional NSAIDs can be used. (B) With increased CV risk, combination of NSAID with low dose ASA is also justified. (C) If GI risks are present, then either a proton pump inhibitor (PPI) should be added or the NSAID should be replaced by a selective COX-2 inhibitor. In the lower GI tract, PPIs do not reduce the (albeit lower) risk of NSAID lesions. (D) With increased CV risk, plain forms of ASA can be used for CV prophylaxis and the predominantly gastroduodenal lesion risk can be reduced by PPIs. At the same time, PPIs reduce the potential of NSAIDs to cause gastroduodenal lesions. The potential of NSAIDs for causing lesions in distal sections of the gut can be combated by replacing NSAIDs with a selective COX-2 inhibitor and, where applicable, sustained-release ASA with plain ASA. The indication for treatment (dose of drug, duration of treatment), GI risk, and CV risk are a basis for the individual risk–benefit evaluation of anti-inflammatory treatment.
REFERENCES
Footnotes
Published Online First 7 June 2005
Disclosure: Dr W W Bolten has received speaker’s fees from Pfizer, MSD, Novartis and AstraZeneca for his presentations.