Article Text

Abstract
Intravenous immunoglobulin (IVIg) is an adjunctive therapy for juvenile dermatomyositis (JDM) patients with poor response to first-line therapy (corticosteroid resistant; SR) or who are corticosteroid dependent (SD). Patients requiring IVIg are generally expected to have poorer outcomes, leading to confounding by indication in observational studies. Few studies have evaluated IVIg efficacy in JDM.
Objectives Compared with similar matched controls, to determine if JDM IVIg recipients achieve quiescence sooner and have less disease activity. For SD patients, to determine if IVIg recipients exhibit less disease activity than IVIg-naive patients.
Methods A retrospective inception cohort of 78 JDM patients was studied. Kaplan–Meier survival determined time to quiescence. Marginal structural modelling was used to account for confounding by indication by incorporating inverse probability of treatment weights to handle patients' unequal likelihood of receiving IVIg.
Results While similar demographically, the 30 IVIg patients demonstrated weaker muscle strength and more had photosensitivity at baseline than the 48 controls. As expected, IVIg patients achieved quiescence later than controls in unadjusted analysis. However, although IVIg patients started with greater disease activity, after accounting for confounding as best possible, they maintained similar or lower disease activity than controls from 30 days to 4 years post-diagnosis. This improvement was most marked in SR patients. Among SD patients, IVIg recipients maintained lower disease activity than IVIg-naive patients.
Conclusion This study, involving the largest JDM cohort receiving IVIg to date, applied bias-reduction methods and demonstrated IVIg efficacy in controlling JDM disease activity, particularly for SR patients.
Statistics from Altmetric.com
Juvenile dermatomyositis (JDM), the most common inflammatory myopathy in children, is a rare but important chronic multisystem disease that often requires long-term therapy.1 2 The aetiology and pathogenesis of JDM remain unclear but JDM is thought to be autoimmune in nature, with therapy based on immunosuppression to reduce inflammation. Corticosteroids (oral or intravenous pulse) are regarded as a standard mainstay therapy worldwide.3
There is great interest in alternative and adjunctive therapies to corticosteroids in JDM. Although many patients respond rapidly to corticosteroids, treatment is often prolonged and there is a high risk of adverse effects, both acute (eg, gastritis) and long term (eg, cataracts, osteoporosis and growth impairment). Methotrexate has been widely used as a corticosteroid-sparing agent,4 but there remain cases refractory to corticosteroid and methotrexate. Intravenous immunoglobulin (IVIg) has been utilised as an adjunctive therapy, typically for two groups of JDM patients: corticosteroid-resistant (SR) patients who do not respond adequately to corticosteroids (and other adjuncts such as methotrexate) early in the disease course and corticosteroid-dependent (SD) patients who respond initially but are unable to be weaned off therapy without disease exacerbations.
IVIg has been utilised in JDM with apparently favourable responses since the 1980s, noted in case reports5,–,7 and small case series.8,–,11 One small controlled clinical trial was conducted with IVIg in adult dermatomyositis, showing improved strength and neuromuscular symptoms in those receiving IVIg.12 A recent survey of North American paediatric rheumatologists suggests that there is particular interest in applying IVIg to manage those with severe/refractory disease and significant skin disease,13 and expert consensus outside of North America similarly supports the consideration of IVIg in such contexts.14 However, JDM is rare, thus appropriately powered randomised trials of IVIg have been considered unfeasible. There are few systematic data to evaluate the efficacy of IVIg for treating JDM.
Evidence has been limited by difficulties of interpreting observational data (with possible biases from concomitant therapies, variable dosing and courses of IVIg) as well as small samples. Most particularly, confounding by indication complicates the interpretation of IVIg efficacy in observational studies; patients prescribed IVIg are usually sicker and expected to have a poorer prognosis and more difficult clinical course. The reason a patient is selected to receive IVIg may have a greater impact on disease outcome than the treatment itself.
Marginal structural modelling has emerged as a means to help establish causal inferences using observational data, by adjusting for baseline confounding biases between treatment and control groups.15 16 A marginal structural model (MSM) was developed in this retrospective cohort study—aiming to account for confounding by indication by incorporating inverse probability of treatment weights to account for patients' unequal likelihood of receiving IVIg.
The goal of this study was, using a retrospective inception cohort, to examine the efficacy of IVIg in JDM using a MSM to account for confounding by indication. First, we used an unadjusted analysis to examine the time to achieve quiescence, when quiescence is defined as having no active disease while on anti-inflammatory and/or immunosuppressive medications, comparing control JDM patients with patients receiving IVIg. After adjusting for confounding by indication through a MSM, we examined whether patients receiving IVIg exhibit less disease activity, using an adapted disease activity score (DAS) based on muscle (MSK) and skin (SKIN) findings.17 Finally, in a subgroup analysis examining SD patients, we determined whether those receiving IVIg maintained less severe disease.
Methods
Population and design
This was a retrospective study with an inception cohort composed of patients followed from diagnosis onward at a dedicated JDM clinic at the Hospital for Sick Children (SickKids). Ethics approval was obtained from the institutional Research Ethics Board.
Since 1991, all patients with JDM presenting to SickKids have been treated at our tertiary subspecialty multidisciplinary clinic with defined protocols and standardised data collection, with all clinical and laboratory data stored in a central database. We identified 135 patients, of which 78 were included in our research cohort, having been diagnosed in our clinic between June 1991 and December 2005. Subjects meeting the initial inclusion criteria were those aged 18 years or younger at the onset of their disease, with a diagnosis of probable or definite JDM according to the Bohan and Peter criteria.18 19 Excluded were patients diagnosed before 1991 (for whom we did not have sufficient data and before which time IVIg had not yet become a common adjuvant therapy), those subsequently given an alternative diagnosis, and those for whom we had inadequate information (having been referred to us after their diagnosis, or with fewer than three visits, or were not treated at our facility). Of the 78 subjects included in our cohort, 48 were control JDM patients who responded to first-line therapy; 30 were recipients of IVIg, of which 14 were SR, with 16 SD (figure 1).
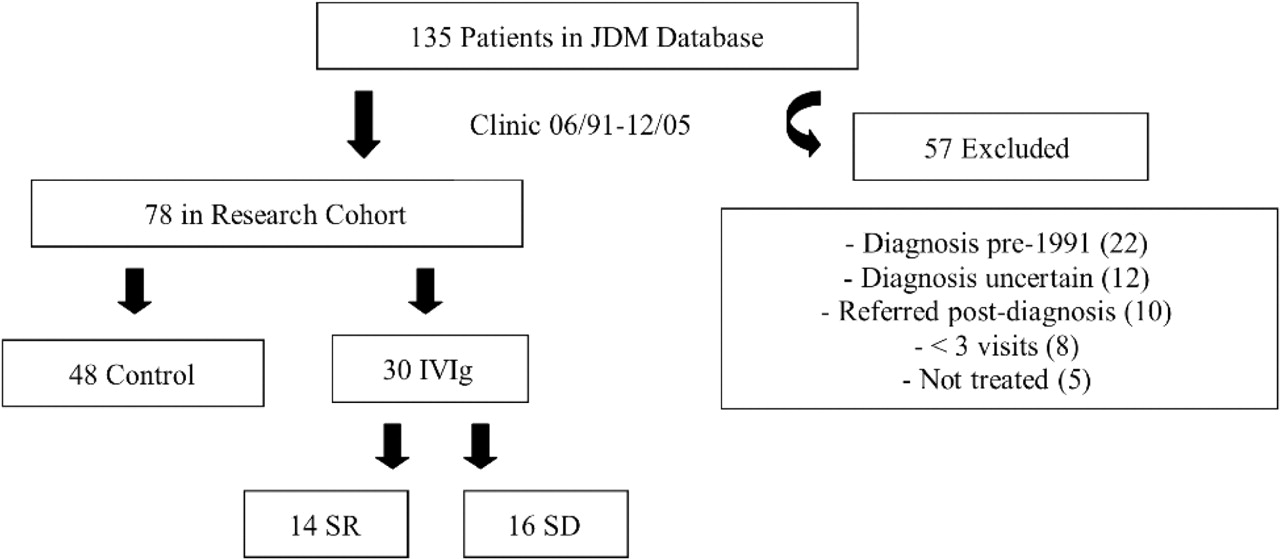
Disposition of patients with juvenile dermatomyositis (JDM) seen at the Hospital for Sick Children (SickKids) during the study period. SD, corticosteroid dependent; SR, corticosteroid resistant.
Therapies
Our standard approach to treating JDM patients is to give high-dose oral corticosteroids for all patients; since 1994 methotrexate has been given to most patients as a corticosteroid-sparing agent. Prednisone was given at 2 mg/kg per day (maximum 100 mg/day), consolidated to a single daily dose 6 weeks after diagnosis; intravenous pulse methylprednisolone at 30 mg/kg per treatment (maximum 1000 mg/dose) was also occasionally used for those with poor response or tolerance to oral steroids. Those not improved at 6 weeks were considered SR. SR was defined as failing to show expected clinical improvement after 6 weeks of standard therapy, as determined by the treating paediatric rheumatologist, considering objective clinical measures (muscle and skin disease activity), patient-reported functional status (child health assessment questionnaire; CHAQ) and laboratory markers when appropriate. Improvement on first-line treatment was determined similarly. In all cases, standardised clinical assessment forms specifically created for the JDM clinic were used to assess whether abnormal or concerning features were present. Of note, included also in the SR group (n=14) were nine patients deemed by the treating rheumatologist to have severe disease at diagnosis, with features such as marked dysphagia or severe weakness, and were thus treated at the outset with IVIg in addition to standard therapy. Those improved on first-line treatment at 6 weeks after diagnosis but who manifested recurrent symptoms upon tapering of the corticosteroids were considered SD. The JDM patients demonstrating maintained improvement on corticosteroid therapy were considered controls in our study (ie, they were not treated with IVIg). Of the 78 patients in our cohort, all but 20 patients also received methotrexate as part of their initial therapy, at 10–20 mg/m2 per week (maximum 25 mg/week). All SD and SR patients received IVIg (Gamimune, Bayer AG, Gammagard, Baxter International Inc or IVEEGAM, Baxter International Inc) as determined by hospital pharmacy supply. Patients were treated with IVIg at 2 g/kg per infusion (maximum 70 g), with IVIg infusions administered every 2 weeks for the first five treatments. If there was a positive response, IVIg was continued monthly for a planned duration of approximately 2 years, to allow for a gradual tapering frequency of infusions (with 6, 8 and then 12 weeks in between infusions) after successful discontinuation of corticosteroid.
Clinical tools and definitions
Quiescence was defined as having no active disease while on therapy, characterised by normal muscle strength, skin, physical function (as determined by the CHAQ when available) (The CHAQ was administered at every visit; due to respondent fatigue, it was not completed by all subjects at each visit. A normal CHAQ was considered to be less than 0.13)20 21 and muscle enzymes (creatine phosphate kinase, aspartate aminotransferase and alanine aminotransferase).
Adapted DAS
This was a 12-point DAS score based on the Chicago DAS validated in Bode et al,17 adapted for retrospective studies, wherein a higher score corresponds to worse disease activity. The adapted score includes a DAS MSK (7 points) and a DAS SKIN (5 points).
DAS MSK
▶ Functional status (maximum 3 points), in which functional limitation is scored as none (0), limited to extra-curricular activities (1), school (2) or self-care (3).
▶ Muscle weakness (maximum 3 points), using adapted manual muscle testing (MMT) scores (MMT scores were re-coded from a 0–5 Oxford grading score to a scale of 1–10 for each of seven standardised motor movements (neck flexion, right shoulder abduction, left shoulder abduction, right hip flexion, left hip flexion, right hip abduction and left hip abduction), and summed together to yield a global MMT that had a scale of 7–70. Global MMT scores of 68–70 were considered normal), in which 68–70=0, 63–67=1, 56–62=2, 55 or less=3.
▶ Arthritis (1 point if present).
DAS SKIN
▶ Erythema (2 points if present).
▶ Gottron's (1 point if present).
▶ Heliotrope (1 point if present).
▶ Vasculitis (1 point if present)—includes abnormal nailfold capilloscopy.
Statistical methods
Kaplan–Meier survival analysis was used to analyse the unadjusted time to disease quiescence. Continuous and categorical variables were assessed for significance using the student's t test and Fisher's exact test, respectively. The analysis of DAS and prednisone over time was done through a MSM applied to a generalised estimating equation (GEE). MSM is a technique to obtain causal inferences from observational data that is especially suitable when treatments are administered in a time-dependent way, and when there are covariates that both confound the treatment–outcome relationship and are on the causal pathway between treatment and outcome. Each data point is weighted by the inverse of the probability of the observed treatment pattern occurring, multiplied by the inverse of the probability of not being censored. Details of our modelling strategy have been published,22 with details specific to our population included in the supplementary appendix 1 (available online only). Patients tended to visit more often when they were unwell. To avoid biased results, a balanced dataset was created in which the mean DAS and prednisone levels for each patient in the time frames of less than 30 days, 30 days to 1 year, 1–2 years, 2–3 years, 3–4 years and 4–5 years were calculated. These were then analysed using GEE with an autoregressive correlation structure and the MSM weights calculated as described.22
Initially, the difference in mean DAS between patients on IVIg versus not on IVIg in each of the six time intervals was estimated using the GEE. The interaction between indication for IVIg (corticosteroid dependence vs corticosteroid resistance) and IVIg use was examined for significance. As this was strongly significant in many time intervals, all remaining analyses considered IVIg for corticosteroid resistance and for corticosteroid dependence separately. As expected, patients on IVIg for corticosteroid resistance tended to start IVIg earlier in the study than patients on IVIg for corticosteroid dependence. Given the concern that SD patients tended to have more severe DAS even early on before starting IVIg, the effectiveness of IVIg in SR patients was assessed by removing patients who would become SD from the cohort, and just comparing SR patients with controls. The GEE model thus included time category, an indicator for whether the patient was in the SR cohort and the SR by time interaction. When assessing the effectiveness of IVIg for corticosteroid dependence, SR and SD patients were used; however, as patients started IVIg at very different times, the time on IVIg was taken into account. For this reason, the GEE model included time category, an indicator for whether the patient was in the SD cohort, the SD by time interaction, and a covariate for time on IVIg for the SD group (<30 days, 30 days to 1 year, 1–2 years, 2–3 years, >3 years). The coefficients for time on IVIg in the SD group thus compare the mean DAS levels among patients on IVIg for SD for a particular length of time with the mean DAS levels among SD patients who are as yet IVIg naive.
Our MSM included specific indicators for whether the patient was on methotrexate, the prednisone dose, and the prednisone by methotrexate interaction, so that concomitant therapies were no longer confounders (see supplementary appendix 1, available online only).
Results
Demographics
There were no statistically significant demographic differences between control JDM patients and IVIg recipients (see table 1). For those in the IVIg group, the 16 SD patients were all female, followed for a longer period of time and diagnosed at a younger age; there were no significant differences between the SD and the SR patients with regard to their ages at onset of IVIg and course of IVIg, with the expected exception that SD patients were followed for a longer time before starting IVIg (see table 1).
Demographic data of all patients
Baseline clinical status
There were significant differences between the control and IVIg patients. Patients receiving IVIg exhibited weaker strength as determined by MMT (especially the SR patients), with a smaller proportion having subjective weight loss, and a higher proportion having photosensitivity (especially the SD patients) (see table 2).
Baseline clinical features of all patients
Treatments
Intravenous pulse methylprednisolone was given to 13 patients (six in the control group, three in the SR group and four in the SD group). IVIg was given at an average rate of one infusion per month (initially more frequently, and less frequently with time), over 30 months, with a range of seven to 88 total infusions. IVIg was generally well tolerated. There were no reports of severe or life-threatening adverse incidents; the most commonly reported adverse events included fever, headache, nausea/vomiting and lethargy, with 9% of infusions resulting in such events. Our experience with IVIg safety has been published previously.23
Analysis
Given that patients are prescribed IVIg because of a difficult clinical course and are generally expected to have a more guarded prognosis, it was not surprising that the unadjusted Kaplan–Meier survival analysis found that control patients achieved quiescence earlier (figure 2).
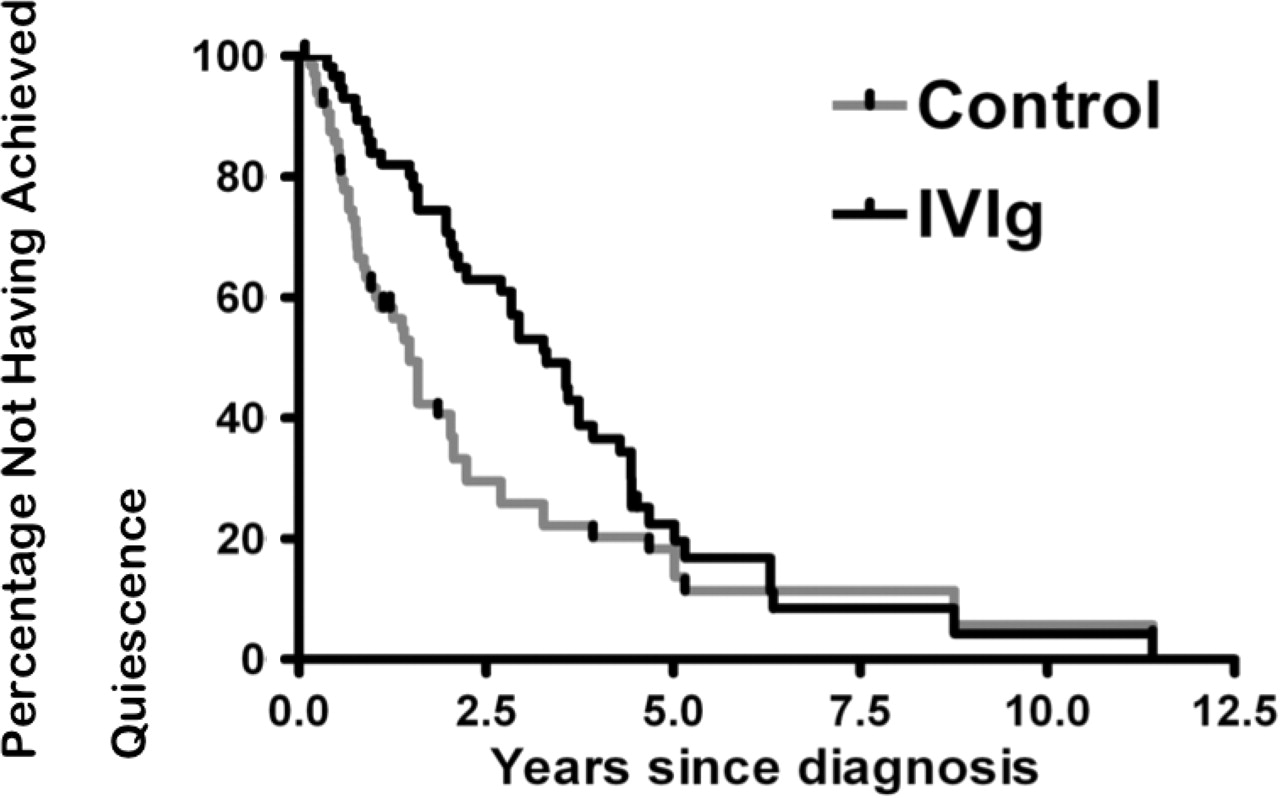
Kaplan–Meier survival: unadjusted time to quiescence.
This confirmed our need for more sophisticated statistical methods to account for possible confounding by indication. Using a MSM to account for confounding, despite starting with more severe disease activity compared with controls, IVIg recipients as a whole from 30 days to 4 years after diagnosis maintained a less active or similar disease activity relative to controls (figure 3). Further analysis demonstrated strong interactions between IVIg indications (ie, whether patients were SD or SR), IVIg use and disease severity, warranting subgroup analyses. Compared with controls, SR patients receiving IVIg demonstrated a significant reduction in DAS, particularly from 30 days to 4 years after diagnosis (figure 3 and table 3).

{kind=link}
{kind=link}
{kind=link}
Control (CR) vs intravenous immunoglobulin (IVIg) on disease activity score (DAS): using a marginal structural model, mean DAS values for patients on IVIg vs those patients not on IVIg, as well as the subgroups of IVIg-receiving patients considered corticosteroid dependent (SD) and corticosteroid resistant (SR) at the time indicated are shown. Note: patients' duration of IVIg therapy is not considered; patients who initiate IVIg are counted as being on IVIg for the remainder of the study. Higher DAS reflects more severe disease activity. Data values are below the figure. Data shown as mean DAS (SEM).
Difference in mean DAS by time interval between those exposed to IVIg vs those currently unexposed, for all IVIg recipients as well for patients grouped by reason for IVIg
In comparing DAS of controls with SD patients, more severe disease activity was persistently noted in SD patients beyond 30 days after diagnosis, despite the use of our MSM (figure 3 and table 3). However, among the SD patient subgroup, those started on IVIg tended to have lower mean DAS throughout follow-up (from 1 year after IVIg exposure) when compared with the SD patients not yet started on IVIg (although not significantly so; see table 3).
Discussion
This study provides preliminary evidence that IVIg is efficacious in severe or refractory cases of JDM, particularly in patients with SR disease. With statistical bias-reduction methods to account for confounding by indication, it was shown that IVIg may bring about better control of disease activity, most notably in SR patients.
The control and IVIg-receiving JDM patients in our sample were, overall, demographically similar to each other, with a relative predominance of females diagnosed in the school-age years, as consistent with the literature.24 The subgroup analysis, however, found exclusively females in the SD group; males have previously been described in other SD samples,11 but it is unclear from the small samples, including the 16 patients in this instance, whether there may exist a genuinely greater female predominance in this subgroup. The significant differences in the baseline characteristics between control and IVIg-receiving patients confirmed the expectation of more severe presentation of IVIg-requiring patients.
Current treatment for JDM typically involves prednisone with methotrexate, with IVIg reserved for refractory cases. Our results support the limited data available to date on the efficacy of IVIg in dermatomyositis in improving strength and neuromuscular symptoms,12 as shown by the decrease or improvement in global DAS in our case. IVIg is used in multiple clinical contexts for its presumed anti-inflammatory therapeutic effects. Various proposed mechanisms for its action include modulation and blockade of immunoglobulin cellular receptors, autoantibody neutralisation, and modulation of cytokine and complement pathways.25 Specific mechanisms proposed for its utility in dermatomyositis have included post-IVIg downregulation of inflammatory cytokines (eg, transforming growth factor beta) and interception of the complement pathway's membranolytic attack complex.26
As therapy-related complications with corticosteroids and other immunosuppressants are well documented, it is noteworthy that IVIg appears generally safe, with no severe adverse events reported, in this study as well as in recent reviews.23 25
This study demonstrated potential particular utility for IVIg in SR patients. While use of the MSM aimed to account for the greater disease severity in IVIg-requiring patients in comparison with controls, modelling limitations persisted. Despite our attempts to control for it, relative to controls DAS remained higher in all IVIg patients, including SR patients initially, as well as in SD patients throughout. This modelling limitation may mean that we have underestimated the extent of IVIg efficacy in JDM (ie, that we have not been successful at fully correcting for confounding by indication).
Our data must be interpreted within the context of potential limitations imposed by our study design—including its retrospective nature; available data did not include the JDM ‘core set’ of measures that were developed over the period of this study.27 However, despite this, our study had the use of rigorously collected data with standardised protocols from a dedicated JDM clinic, with all patients being closely followed as an inception cohort from diagnosis. The small size of this study sample may also preclude definitive conclusions. This was a small study (although the largest of its type to date) and therefore may suffer from low statistical power to detect efficacy; the fact that we found positive results in many of our analyses suggests a real effect of IVIg. Finally, although this study made use of an observational cohort, in which confounding by indication is expected, our use of statistical methods for bias-reduction is a strength, and highlights a technique that may prove useful in other observational studies in rare rheumatic diseases. Certainly, our study supports the use of this or similar methods of bias reduction in the study of larger cohorts of IVIg-treated patients with JDM.
In conclusion, our findings support the efficacy of IVIg in controlling disease activity in severe or refractory JDM, particularly in patients demonstrating severe disease at diagnosis or who exhibit SR disease soon after diagnosis. It is hoped that these findings will help guide future practice. In addition, our successful use of bias-reduction methods highlight the utility of statistical models that enable adjustment for confounding biases with observational data in settings where randomised clinical trials are unfeasible.
References
Supplementary materials
Web Only Data
Files in this Data Supplement:
Footnotes
-
Competing interests None.
-
Ethics approval This study was conducted with the approval of the institutional Research Ethics Board of the Hospital for Sick Children, Toronto, Ontario, Canada.
-
Provenance and peer review Not commissioned; externally peer reviewed.