Article Text

Abstract
Objectives: To compare the prevalence and pattern of neuropsychiatric (NP) syndromes observed in systemic lupus erythematosus (SLE) to patients with Primary Sjögren syndrome (PSS) using the American College of Rheumatology (ACR) criteria for the 19 NP syndromes seen in SLE.
Methods: A population-based study was conducted including 68 patients with SLE (mean (SD) age 43.8 (13.6) years) and 72 with PSS (age 57.8 (13.0) years). Specialists in internal medicine, neurology and neuropsychology performed standardised examinations. Cerebral MRI scans and neurophysiological studies were performed in all patients.
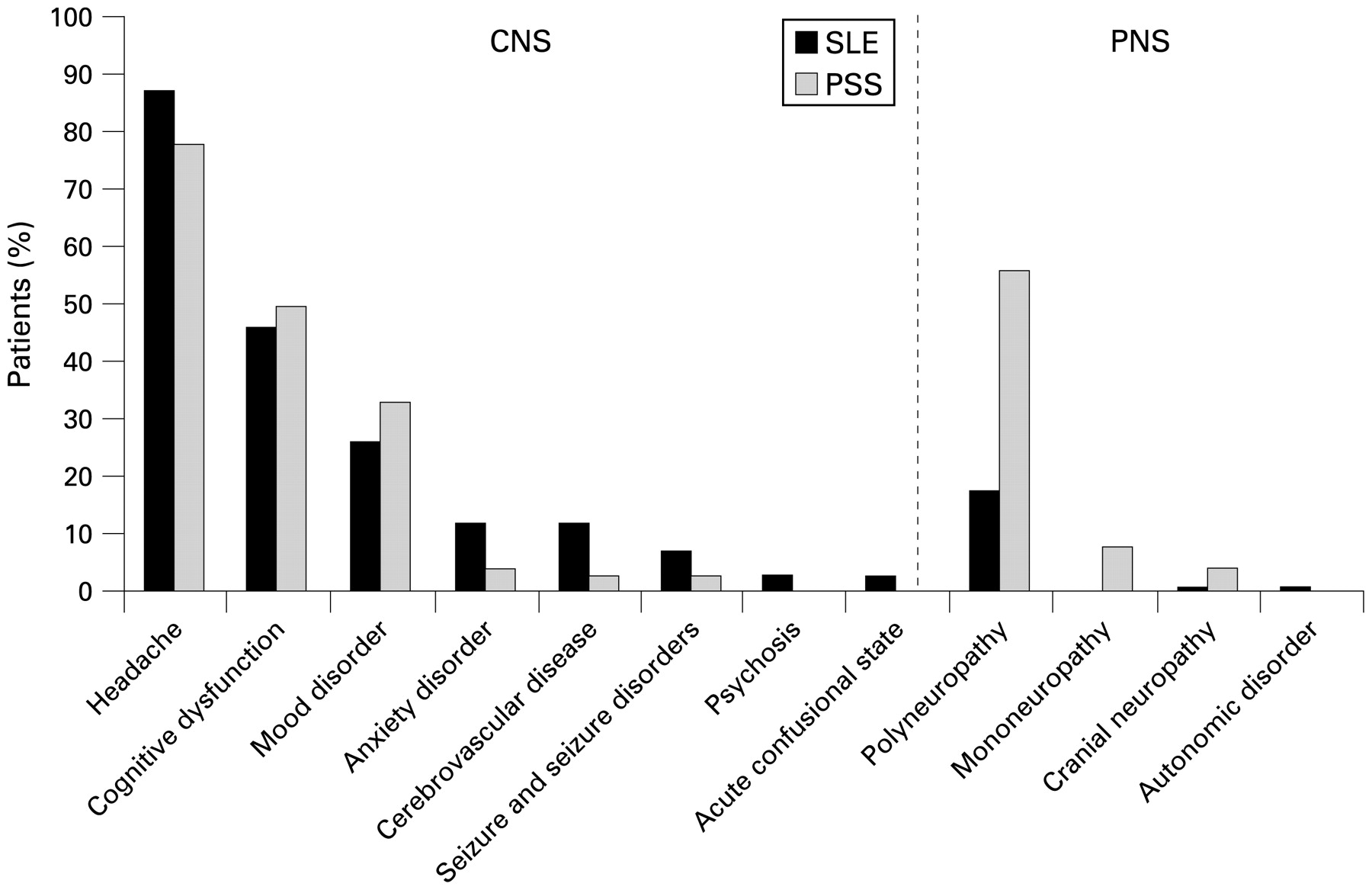
Results: Similar prevalences in SLE and PSS were observed for headaches (87% vs 78%, p = 0.165), cognitive dysfunction (46% vs 50%, p = 0.273), mood disorders (26% vs 33%, p = 0.376), anxiety disorders (12% vs 4%, p = 0.095), cranial neuropathy (1% vs 4%, p = 0.339) and seizure disorders (7% vs 3%, p = 0.208). Cerebrovascular disease was more common in SLE than PSS (12% vs 3%, p = 0.049); but mononeuropathy (0% vs 8%, p = 0.015) and polyneuropathy (18% vs 56%, p<0.001) were less common in SLE than PSS. Other syndromes were rare or absent in both patient groups.
Conclusions: Headache, cognitive dysfunction and mood disorders are common in both diseases, but otherwise there are distinct differences in NP involvement, with cerebrovascular diseases more prevalent in SLE and neuropathies more common in PSS. This indicates that some NP disease mechanisms are shared while others differ between the two diseases.
Statistics from Altmetric.com
The criteria and case definitions proposed by the American College of Rheumatology (ACR) for the neuropsychiatric systemic lupus erythematosus (NP-SLE) syndromes have become widely accepted and cover a wide range of NP manifestations seen in patients with SLE.1
SLE and primary Sjögren syndrome (PSS) share several immunological and clinical features, including involvement of the nervous system. While the impact of SLE on the central and peripheral nervous system is well documented, research on the NP aspects has been hampered by shifting diagnostic criteria for PSS. The recent European–American consensus criteria facilitate agreement on diagnosis of PSS and optimally exclude other causes for sicca phenomena. These criteria emphasise the need for objective measurements, including a salivary gland biopsy with infiltrating mononuclear lymphoid cells, the detection of anti-Sjögren syndrome-associated antigen A (SSA) or anti-SSB antibodies, and certain other objective and subjective phenomena.2
The aim of this population-based study was to apply the NP-SLE diagnostic criteria to patients with PSS that fulfil the European–American criteria, and to compare the prevalence and pattern of NP involvement between patients with PSS and those with SLE. Similarities would indicate that mechanisms for NP manifestations are shared, and differences would indicate that pathophysiological and pathogenetic processes differ between the two diseases.
Methods
This study was approved by the regional research ethics committee and carried out in compliance with the Helsinki Declaration. Stavanger University Hospital is the only local hospital in the southern part of Rogaland county, Norway and provides services to about 300 000 inhabitants. In this region all patients with SLE and PSS are allocated to Stavanger University Hospital. All patients were examined in the hospital over a 2-day period; examinations consisted of blood and urine sampling, review of the medical history, and clinical examinations by specialists in neurology (ABT), internal medicine (EH) and neuropsychology (SM). After discharge the patients had MRI examinations within 1–3 weeks. All patients were examined between March 2003 and March 2006. Establishing a diagnosis of a past or current NP syndrome was based on medical history, clinical interview, current findings and relevant laboratory investigations. The number of NP syndromes reported were the accumulated events in each patient. In consensus meetings among the investigators, all patients were classified according to the standardised ACR NP-SLE nomenclature and case definitions applying the recommended diagnostic criteria, exclusions and methods for ascertainment.1
Patients with SLE
Medical records were reviewed of all inpatients and outpatients diagnosed as having SLE between 1980 and 2004 at Stavanger University Hospital, Norway. In all, 86 patients, all Caucasians, fulfilled the revised ACR criteria for SLE.3 4 Of the 86 patients, 68 (79%) gave informed consent to participate. Patient characteristics, current medication and laboratory findings are presented in tables 1 and 2.
Demographic and clinical characteristics, and use of medication in 68 patients with systemic lupus erythematosus (SLE) and 72 patients with primary Sjögren syndrome (PSS)
Laboratory tests in patients with systemic lupus erythematosus (SLE) and primary Sjögren syndrome (PSS)
Disease activity was measured with the SLE Disease Activity Index (SLEDAI), and organ damage was measured with the Systemic Lupus International Collaborating Clinics/ACR damage index (SLICC).5 6
Patients with PSS
As for patients with SLE, medical records were reviewed of all inpatients and outpatients diagnosed as having PSS between 1980 and 2005 at the Stavanger University Hospital, Norway. Of the 99 patients that fulfilled the European–American criteria for PSS,2 72 patients (73%) gave informed consent to participate. Patient characteristics, current medication and laboratory findings are presented in tables 1 and 2.
Neuropsychological testing
Neuropsychological testing mapped functions in the following eight cognitive domains: simple attention, complex attention, memory, visual–spatial processing, language, reasoning/problem solving, psychomotor speed and motor function. The tests were administered by a trained psychometric test technician starting at a fixed time in the morning for all patients. The neuropsychological test battery included: Trail Making Test A, Trail Making Test B, Category Test, Seashore Rhythm Test, Lafayette Hand Dynamometer Test, Finger Tapping Test, Tactual Performance Test, Lafayette Grooved Pegboard Test,7 Wechsler Adult Intelligence Scale (WAIS),8 Wechsler Memory Scale Revised,9 Wisconsin Card Sorting Test,10 FAS Verbal Fluency test,11 and Stroop Colour–Word Interference Test.12 Subtests from the WAIS test were used to estimate premorbid levels of function. The scores were compared to the normative data for each test.7 9 10 11 13 14 15 16 17 18 19 20 A cut-off score for abnormality was defined as a standardised score ⩾2 SD from the reference mean.
Grading of cognitive function
Cognitive dysfunction was classified as mild if there were deficits in less than three domains, moderate if there were deficits in three or four domains and severe if there were deficits in five or more domains.21 Moderate and severe dysfunction both imply impairment in everyday functioning.1 The tests were completed within 3–4 h.
Cerebral MRI
Patients were examined by MRI in a 1.5-T Philips Gyroscan NT Intera Release 10 (Philips Medical Systems, Best, The Netherlands). A total of 63 (93%) of the patients with SLE and 69 (96%) of the patients with PSS completed an MRI examination. The patients with SLE were examined at 12.4 (11.4) days and patients with PSS were examined at 13.6 (8.0) days after the clinical examination.
Nerve conduction studies (NCS)
A total of 56 (82%) of the 68 patients with SLE and 70 (97%) of the 72 patients with PSS completed NCS, which was performed under standard conditions as previously described.22 The neurophysiological criterion for polyneuropathy was defined as abnormalities in two or more nerves.23
Measurements of quality of life, fatigue and mood
The Short Form-36 (SF-36) was used for measurements of self-reported quality of life.24 The Fatigue Severity Scale (FSS) and a fatigue visual analogue scale (VAS) were used for fatigue measurements, and the Beck Depression Inventory (BDI) was used to assess mood.25 26 A BDI cut-off score of ⩾13 is commonly used to identify current clinical depression.27 The diagnosis of current or past mood disorder was based on the medical history, clinical interview, current treatment for depression and BDI scores.27
Laboratory tests
Routine haematological and biochemical tests were analysed in the hospital laboratory. Anti-nuclear antibodies (ANA) were detected by a HEp-2000 assay (Immunoconcepts, Sacramento, California, USA), and antibodies to double-stranded DNA (dsDNA) were verified by a Nova Lite dsDNA Crithidia luciliae 708200 indirect immunofluorescence assay (Nova Diagnostics, San Diego, California, USA). Screening for SSA/Ro and SSB/La autoantibodies, was performed with the QUANTA Lite ENA 6 (Inova Diagnostics, San Diego, California, USA), and positive results were confirmed by the QUANTA Lite SSA and SSB ELISA (Inova Diagnostics). Screening for anti-cardiolipin IgM and IgG antibodies was performed with the QUANTA Lite ACA IgM and IgG ELISA (Inova Diagnostics). Lupus anticoagulant (LA) was screened for using activated partial thromboplastin time and dilute Russell’s viper venom time (Dade Behring, Marburg, Germany). Complement factors C3 and C4 were measured by nephelometry.
Statistical methods
Results are reported as the mean (SD) when normally distributed, otherwise as the mean (SD) with median and range. Data from patients with PSS and SLE measured on a continuous scale were compared using t tests when normally distributed and the Mann–Whitney U test otherwise. The χ2 test was applied for comparing categorical data. When analysing ordered categories, we applied the χ2 test for trends. Multiple linear logistic regression was applied to examine the joint influence of different factors on NP syndromes. A significance level of p<0.05 was used.
Results
One or more NP syndromes were found in nearly all of the patients (patients with SLE 66/68 (97%) vs patients with PSS 69/72 (96%); p = 0.696) (fig 1).

Number of neuropsychiatric (NP) syndromes observed in each patient with systemic lupus erythematosus (SLE; n = 68) or primary Sjögren syndrome (PSS; n = 72).
Among the 68 patients with SLE, 27 (40%) had 1 NP syndrome, 12 (18%) had 2, 17 (25%) had 3, 8 (12%) had 4, 1 had 5 and 1 had 7. Among the 72 patients with PSS, 17 (24%) had 1 NP syndrome, 17 (24%) had 2, 23 (32%) had 3, 10 (14%) had 4 and 2 (3%) had 6. Trend analysis showed there were no differences among patient groups when comparing the number of NP criteria observed (p = 0.296), fig 2.
{kind=link}
{kind=link}
Distribution of neuropsychiatric (NP) syndromes observed in patients with systemic lupus erythematosus (SLE; n = 68) or primary Sjögren syndrome (PSS; n = 72). Guillain–Barré syndrome, aseptic meningitis, demyelinating syndrome, movement disorder, myasthaenia gravis, myelopathy and plexopathy were not observed in either patient group. CNS, central nervous system; PNS, peripheral nervous system.
NP syndromes excluding headache showed a prevalence of 63% in the patients with SLE versus 75% in the patients with PSS p = 0.132.
Autoantibodies and NP syndromes
When considering NP syndromes combined (excluding headache), there were no associations between the presence of anti-DNA, anti-SSA, anti-SSB or anti-phospholipid antibodies and NP syndromes. Additionally, when separately evaluating the most prevalent phenomena (ie, cognitive dysfunction, mood disorder and polyneuropathy), there were no associations between these antibodies and the individual NP syndromes except for anti-SSA antibodies and cognitive dysfunction in patients with PSS (32 (89%) patients with PSS with vs 25 (69%) patients with PSS without cognitive dysfunction, p = 0.042).
Cerebrovascular disease
Eight patients with SLE and two with PSS had cerebrovascular disease. Among the patients with SLE, six had experienced a stroke and two a stroke and a transient ischaemic attack. Among the patients with PSS, one had experienced a stroke and one a transient ischaemic attack. More patients with SLE than with PSS were positive for anti-phospholipid antibodies (aPL) (ie, the presence of anti-cardiolipin IgM antibodies (aCL IgM), anti-cardiolipin IgG antibodies (aCL IgG), or lupus anticoagulant (LAC); table 2). However, only four of the eight patients with SLE and cerebrovascular disease were positive for aPL.
Cognitive function
Cognitive impairment was common in both patient groups and did not differ in prevalence (table 3); nor were any differences evident when comparing severity (grading) of cognitive dysfunction (p = 0.387) by trend analysis. Among the 68 patients with SLE, 37 (54%) had normal cognitive function, 15 (22%) had mild, 9 (13%) had moderate and 7 (10%) had severe cognitive dysfunction. Among the 72 patients with PSS, 36 (50%) had normal cognitive function, 17 (24%) had mild, 15 (21%) had moderate and 4 (6%) had severe cognitive dysfunction.
Neuropsychiatric syndromes in patients with systemic lupus erythematosus (SLE) and primary Sjögren syndrome (PSS)
Headache
Headache was the most prevalent neuropsychiatric manifestation in SLE and PSS, but did not differ between groups (table 3); 30 (44%) of the patients with SLE and 25 (35%) of the patients with PSS had migraines (p = 0.255), while 40 (59%) of the patients with SLE and 40 (56%) of the patients with PSS had tension type headaches (p = 0.696). Cluster headaches, headaches from intracranial hypertension, or intractable headaches were not found in either patient group.
Mood
There were no differences in mood disorders (table 3). Manic features or mixed mood disorder with manic features were not observed in either patient group.
Polyneuropathy
Polyneuropathy diagnosed by NCS or clinical examination, was considerably more prevalent in patients with PSS than those with SLE (table 3). This difference remained significant (PSS vs SLE; p = 0.030) after adjusting for the age difference with multiple linear logistic regression.
Among the 12 patients with SLE and polyneuropathy, 2 cases were diagnosed based on clinical examination, 3 by the combination of clinical examination and NCS findings and 7 by NCS without clinical signs of polyneuropathy. In the 10 patients with abnormal NCS, 5 patients had sensory polyneuropathy, 4 patients sensorimotor polyneuropathy and 1 patient had pure motor polyneuropathy. The patients with SLE and polyneuropathy were significantly older than the patients with SLE without polyneuropathy (62.7 years vs 39.8; p<0.001).
Among the 40 patients with PSS and polyneuropathy, 7 cases were based on clinical examination, 9 by the combination of clinical examination and NCS findings and 24 by NCS criteria without clinical findings. In the 33 patients with abnormal NCS, 9 patients had sensory polyneuropathy, 10 patients had sensorimotor polyneuropathy and 14 patients had pure motor polyneuropathy. The patients with PSS and polyneuropathy were older than the patients with PSS without polyneuropathy (61.5 years vs 53.1 years; p = 0.006).
Mononeuropathy
Six patients with PSS had mononeuropathy or multiple mononeuropathy. None of the patients with SLE had mononeuropathy
Fatigue, depression and quality of life measures
Fatigue, depression and lowered self-reported quality of life were more pronounced in patients with PSS than those with SLE (table 4).
Mood, fatigue and quality of life measures in patients with systemic lupus erythematosus (SLE) and primary Sjögren syndrome (PSS)
Discussion
The main finding of this study was that the NP syndromes were as common in PSS as in SLE. However, the pattern of involvement differed in that cerebrovascular disease was more prevalent in patients with SLE and peripheral nerve disease was more common in patients with PSS. Of particular interest, Cognitive dysfunction was as prevalent in the patients with PSS as in those with SLE. Of note, we also found that and the severity of cognitive dysfunction was similar in both patient groups.
Neuropsychiatric manifestations have been well recognised in SLE, but less attention has been given to the neurological manifestations of PSS. A particular problem in PSS has been the shifting diagnostic criteria for the disease over time, a fact that hampered comparative studies across demographic boundaries and longitudinal studies. The reported prevalences of NP syndromes in patients with PSS in previous studies therefore varies widely from 0% to 100% of patients.28
The reported prevalences of NP syndromes in patients with SLE varies from 28% to 91% between studies that applied the NP-SLE criteria.21 29 30 31 A fundamental problem with the NP-SLE criteria is that several of these criteria have high sensitivity.1
Patients with SLE had more cerebrovascular disease than patients with PSS even though the former cohort was younger than the latter. This is in agreement with several recent studies that demonstrate an accelerated atherosclerosis (carotid plaque) in SLE, most likely caused by multiple atherogenic factors, including immunologically-mediated thrombotic factors, high lipid profiles and corticosteroid use.32 There were more patients with SLE with aPL antibodies compared to patients with PSS while hypertension was similar in the two groups. Regrettably, our information on smoking status and lipids were incomplete and could therefore not be compared, but the difference could indicate that patients with PSS do not share the atherogenic mechanisms that accompany SLE.
Cognitive dysfunction occurred in half of the patients with PSS and SLE, an observation not previously documented in patients with PSS.33 However, previous studies on PSS did not employ the comprehensive battery of tests employed here, and they did not include neuropsychological testing of all patients with PSS.
In current studies on SLE, several pathophysiological mechanisms for cognitive and emotional disturbances have recently emerged. Among these, aPL antibodies, anti-ribosomal P protein antibodies and antibodies to the NR2 subgroup of N-methyl-d-aspartate (NMDA) receptors are potential candidates for playing key roles.34The similarities we found in cognitive NP patterns in SLE and PSS suggest that some mechanisms may be shared by both diseases, but because aPL occur in significantly fewer patients with PSS than patients with SLE, these antibodies cannot be a major contributor to cognitive dysfunction in PSS.
Whether headache is a true neuropsychiatric manifestation of SLE or just a reflection of the high prevalence of headaches in the age groups that cohorts of patients with SLE and PSS belong to is not clarified.35 Our findings show that headache is also a common feature in patients with PSS.
The clinical information of past mood disorders was, in most cases, insufficient to distinguish between major depressive-like episodes and mood disorders with depressive features. Thus, a subgroup analysis of the severity of depressive disorders was not justified.
We found that the peripheral nervous system (PNS) was more affected in patients with PSS than those with SLE. In accordance with a previous study that included some of the patients with PSS studied here, the pattern was dominantly subclinical motor polyneuropathy detected on NCS.22 In contrast, the patients with SLE had a dominance of sensory neuropathy; this suggested that different parts of the PNS were affected in these two diseases, probably reflecting different pathogenetic mechanisms.
According to the ACR criteria, polyneuropathy can be diagnosed by clinical manifestations, findings on NCS, or a combination of both. However, the symptoms and clinical findings are often too vague and unspecific, and a diagnosis of polyneuropathy should be confirmed by NCS.
Quality of life as measured by SF-36 was significantly affected in both diseases compared to normative data.24 Additionally, both diseases showed a profound impact on fatigue measures in accordance with previous studies.36 37 The underlying mechanisms that mediate fatigue and reduce the quality of life are largely unknown.
Some of the NP syndromes were not observed in either patient group (table 3). In contrast, these syndromes were more frequently observed in a retrospective study of patients referred to Neurological or Internal Medicine departments and therefore selected for a high probability of neurological phenomena.33 Of note, we observed no patients with PSS with multiple sclerosis-like disorders, in contrast with the study by Alexander et al.38
The diagnosis of anxiety was based on the clinical interview by the neuropsychologist and on the medical history. A specific instrument for measuring anxiety was not applied. Additionally, we did not have cell counts or protein measures from the cerebrospinal fluid, thus we may have underestimated the prevalence of aseptic meningitis. Further, we did not conduct provocative tests with patients who were asymptomatic to reveal symptoms of autonomic disorders.
The prevalences of anti-DNA, anti-SSA, anti-SSB or anti-phospholipid antibodies were not increased in the patients with SLE or PSS with NP syndromes except for a weak association between anti-SSA antibodies and cognitive dysfunction in patients with PSS. This may be incidental.
There was an age difference between the patients with SLE and those with PSS. However, the cut-off for abnormality in cognitive testing and NCS were adjusted and compared to normative data, thus the age difference cannot explain the findings.
We defined the disease duration to be the time since diagnosis. Several patients were first diagnosed as having PSS at inclusion in this study; thus, the impact of disease duration on different NP syndromes could not be compared.
It is a strength of this study that all patients were diagnosed as having PSS by the recent European–American consensus criteria.2 A study applying four different classification criteria sets to patients with suspected Sjögren syndrome found that different patients fulfilled different PSS criteria sets ranging from 9% to 36% of the patients referred.39
Another strength is the population-based design, thus not biased by patient selection. Additionally, nearly all patients completed the objective testing, including MRI and NCS, regardless of symptoms; in addition, all patients completed the neuropsychological testing. However, the number of patients included was not large enough to evaluate the syndromes that were rare or absent in these patient groups.
Lack of associations between autoantibodies and NP syndromes could be due to the size of this study. Additionally, important molecules such as anti-ribosomal P protein, anti-NMDA receptor or anti-ganglioside antibodies were not analysed.40
In summary, NP involvement in SLE and PSS is common, but the pattern of involvement differs somewhat between the diseases. Future research may take these differences into account when in search of NP syndrome mechanisms.
REFERENCES
Footnotes
Funding EH received support as a doctoral research fellow from the Norwegian Foundation for Health and Rehabilitation.
Competing interests None.
Ethics approval This study was approved by the regional research ethics committee and carried out in compliance with the Helsinki Declaration.