Article Text

Abstract
Background: Antigen-presenting cells (APC) and T cells are considered to play a significant role in the pathogenesis of rheumatoid arthritis (RA). CCL18 and CXCL16 are two chemokines that facilitate T cell attraction by APC, of which a role in the pathogenesis of RA has been suggested.
Objective: To compare the circulating levels of CXCL16 and CCL18 in RA with controls and to investigate the relation of CXCL16 and CCL18 with RA disease activity and joint damage.
Methods: Circulating CCL18 and CXCL16 levels were determined in 61 RA patients with a follow-up of 6 years and a group of 41 healthy controls with ELISA. Chemokine levels were correlated with demographic data, disease activity (DAS28) and joint damage (modified Sharp score). In addition, serum CCL18 and CXCL16 levels from a cohort of 44 RA patients treated with anti-TNF-α were correlated with disease activity.
Results: CCL18 levels in serum were significantly elevated in RA patients compared with controls, while serum CXCL16 levels were not. In contrast to CXCL16, serum CCL18 was positively correlated with disease activity. Both CCL18 and CXCL16 levels decreased upon treatment with anti-TNF-α. Neither CCL18 nor CXCL16 correlated with joint damage and progression.
Conclusion: Here, we show, for the first time, that circulating CCL18 and not CXCL16 levels are elevated in RA patients as compared with controls and correlate with disease activity in RA. More knowledge regarding the regulation and function of both CCL18 and CXCL16 is essential to value their role in RA.
- CCL18
- CXCL16
- disease activity score (DAS-28)
- rheumatoid arthritis (RA)
- TNF-α neutralisation
Statistics from Altmetric.com
INTRODUCTION
Rheumatoid arthritis (RA) is a chronic auto-immune disease, characterised by an inflammation of the synovial joints that eventually leads to cartilage damage and bone destruction. Despite extensive research, the exact pathogenesis of RA is still unclear. Nowadays, there is substantial evidence supporting a role for antigen-presenting cells (APC), such as dendritic cells (DC) and macrophages (MΦ) in RA.1–3 These APC activate T cells and subsequently play a pivotal role in orchestrating immune responses.4 In addition, upon stimulation by T cells, APC act as downstream players in RA and secrete cytokines such as TNF-α and IL-1β, which are now successfully targeted in the clinic.5 6 In order to direct T cell responses, APC first need to attract different T cell subsets. This chemo-attraction is mediated by chemokines (CK). CK constitute a large family of proteins that all possess chemo-attractive capacities towards leucocytes. A subset of the CK family preferentially attracts T cells and is therefore critical in the direction of T cell-driven immune responses.
Recently, we started investigating the role of DC and a large panel of T cell attraction chemokines secreted by those DC in RA, of which CXC chemokine ligand 16 (CXCL16) and CC Chemokine ligand 18 (CCL18) were identified as particularly interesting subjects.7–11 CXCL16 is a unique trans-membrane CK that exists in both a membrane bound and soluble form. Membrane bound CXCL16 is a scavenger receptor for oxidised low-density lipoproteins,12 can facilitate cell adhesion13 and mediates phagocytosis of bacterial fragments.14 This membrane-bound CXCL16 is expressed on APC9 15 16 and can be cleaved by proteases such as ADAM-1017 18 to serve as a chemo-attractant for CXCR6+ cells in its soluble form.16 19 The receptor CXCR6 is present on activated, memory-type T cells, plasma cells and NKT cells.19–21 We recently demonstrated the abundant expression of CXCL16 and CXCR6 in RA synovial tissue and fluid, as well as its regulation by synovial fluid and TNF-α.9 In addition, we found high levels of cleaved CXCL16 in the synovial fluid of RA patients. Recently, additional evidence for a role for CXCL16 in RA was provided as CXCL16 was suggested as a potentially novel therapeutic target in RA as blockade of CXCL16 resulted in a decrease in arthritis in murine collagen-induced arthritis.22
CCL18 (DC-CK1, PARC, AMAC-1) is another T cell attracting CK that was first identified as a chemoattractant for naïve T cells and is produced by DC and alternatively activated MΦ.23–26 CCL18 was initially found in high quantities in the lung (alveolar MΦ) in health and disease.24 Interestingly, a high CCL18 expression was found in synovial tissue of patients with RA,7 27 which suggested that CCL18 might play a role in the pathogenesis of RA. In addition, CCL18 can also act as a profibrotic factor in the lung,28 indicating that T cell attraction is not the only function of CCL18. Circulating CCL18 has been shown to be useful as a biomarker for Gaucher’s disease.29 Moreover, associations with circulating CCL18 levels have been suggested in a large variety of diseases, including lymphoblastic leukemia,30 atopic dermatitis,31 ovarian carcinomas32 and allergic asthma.33
Sensitive biomarkers for disease activity and progression in RA are currently still lacking. The demand for such markers, however, is increasing, since novel therapeutic strategies are very expensive, have serious side effects and vary in efficacy between individual patients. This, in combination with the suggested roles of CXCL16 and CCL18 in RA pathogenesis, prompted us to investigate their circulating levels in RA and their potential correlation with clinical disease parameters. In the present study, we show that serum levels CCL18 are elevated in RA and correlate significantly with disease activity parameters in two independent cohorts, whereas CXCL16 did not show any correlation with disease activity. Neither of the two CK correlated with radiological progression. These results suggest that serum CCL18, but not CXCL16, might reflect the disease course of RA.
METHODS
Patients
Patient serum samples were repetitively taken from 61 patients enrolled in the RA inception cohort of the Radboud University Nijmegen Medical Centre. All patients fulfilled the ACR criteria for the diagnosis RA.34 The first sample was taken at the time of diagnosis, prior to the initiation of treatment with disease-modifying antirheumatic drugs (DMARDs), and is referred to as baseline sample throughout the manuscript. None of the patients had been treated with anti-TNF-α during the period of follow-up we analysed in our study. Patients were seen on a regular basis, and data were collected every 3 months during the first 2 years and every 6 months thereafter. In this cohort, serum samples can be correlated with clinical data from the same day of the blood sample, such as disease activity (DAS-28 score35), joint damage (modified Sharp score36) and laboratory values. Serum samples of healthy volunteers (n = 41) were used as controls to compare CK levels with RA serum levels. In addition, serum samples were taken from 44 patients who where treated with anti-TNF-α (infliximab) in the St. Maartenshospital. The first sample was taken before the first infusion, and the next samples were taken after 2, 6, and 14 weeks. The ESR was measured on the same time points, as were the joint scores and VAS for the DAS-28 score.
Enzyme-Linked Immunosorbent Assay (ELISA)
For the detection of chemokine protein levels of CXCL16 and CCL18 in serum, sandwich ELISAs were performed as described previously.9 37 As an internal control for inter-assay variability, a sample of pooled normal human serum (n = 300) was taken along in all assays. The detection limits for the ELISAs is 100 pg/ml for both CCL18 and CXCL16. The maximum inter-assay variability is estimated at 10%. In order to minimise the effects of this variability, samples that were directly compared were measured in the same assay.
Statistical analysis
In order to evaluate whether circulating chemokine levels were different in RA patients prior to DMARD treatment compared with healthy controls, baseline serum samples from patients were compared with control serum samples with a Mann–Whitney U test. When examining the relation between circulating chemokine levels with disease activity and joint damage, correlations between chemokine levels and clinical data were determined with Pearson’s correlation. CCL18 and modified Sharp scores were root-transformed. To assess the differences between chemokine levels at different time points in our cohort of patients treated with anti-TNF-α, a comparison between baseline and follow-up data was done with a Wilcoxon’s signed rank test. The correlation between changes in DAS-28 and changes in CCL18 or CXCL16 levels was done with a Spearman’s correlation test.
RESULTS
Circulating CCL18 but not CXCL16 levels are elevated in RA
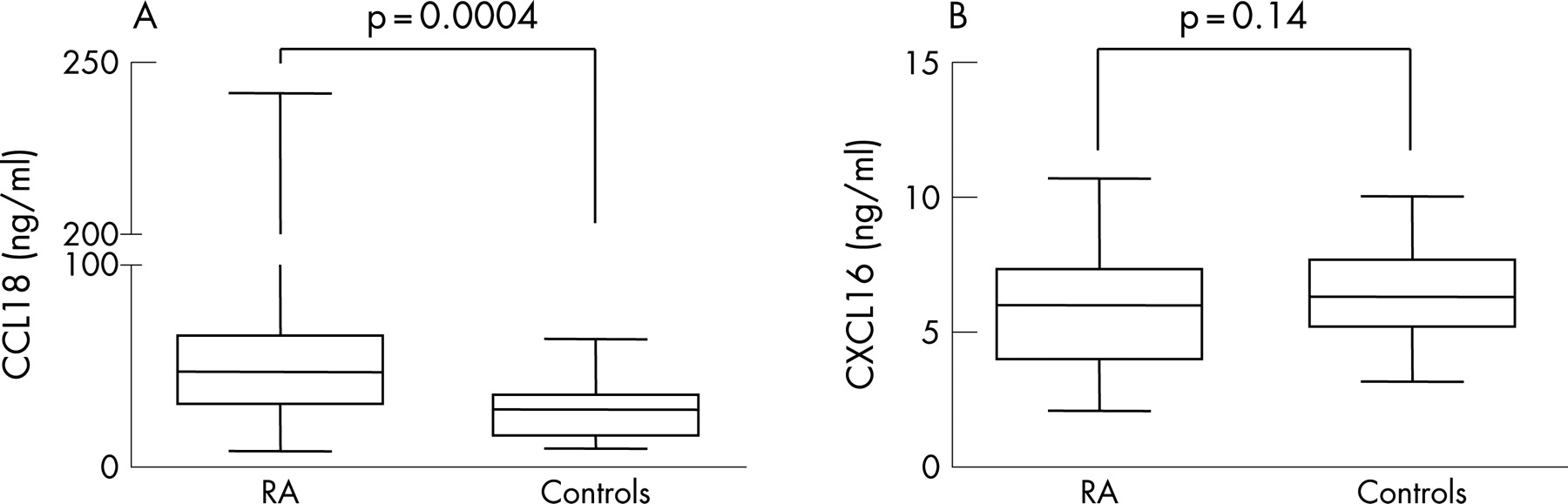
CCL18 and CXCL16 levels prior to DMARD treatment were measured in serum of 61 patients and compared with serum of healthy controls (n = 41). At baseline, CCL18 levels were significantly higher in RA patients (median (interquartile range) 49.0 ng/ml (31.5–71.0)) compared with CCL18 levels in controls (28.6 ng/ml (15.8–35.7)) (p = 0.0004) (fig 1a). In contrast, CXCL16 levels were not significantly elevated in RA patients (6.0 (4.0–7.3) ng/ml) compared with healthy controls (6.3 (5.2–7.7) ng/ml) (p = 0.14) (fig 1b). Neither CCL18 nor CXCL16 levels were associated with demographic or patient characteristics, including rheumatoid factor positivity, age at disease onset and gender as calculated by univariate analysis (data not shown).

CCL18 but not CXCL16 correlates with the disease activity score (DAS-28)
Since we were interested in the relation with disease severity and potential suitability of CCL18 and/or CXCL16 as a biomarker for disease severity in RA, we investigated whether circulating levels of these CK correlated with clinical parameters at baseline and over 3 and 6 years. In table 1, patient and disease characteristics are shown at baseline and over time for progression of joint damage.
SqrtCCL18 at baseline was positively correlated with the DAS-28 score (R = +0.38 (p = 0.003)) and ESR (+0.39 (p = 0.003)) (upper panel of table 2). In contrast, baseline serum CXCL16 levels did not correlate with disease activity. As could be expected, considering their regulation, CCL18 and CXCL16 were inversely correlated, but this correlation did not reach statistical significance (R = −0.22 (p = 0.10)).
Over time, a trend towards a positive correlation between mean serum CCL18 and mean DAS-28 could be observed, although it was less strong compared with baseline (R = +0.21 (p = 0.10) over 3 years R = +0.23 (p = 0.07) over 6 years) (upper panel of table 3). As for baseline, mean CXCL16 over time did not correlate with mean DAS-28 (R = +0.06 (p = 0.63) and +0.02 (p = 0.86) over 3 and 6 years, respectively). Mean CXCL16 and mean CCL18 again showed a trend towards an inverse correlation (R = −0.21 (p = 0.10) and −0. 20 (p = 0.12) over 3 and 6 years, respectively).
Serum chemokine levels do not correlate with joint damage and progression
Cartilage and bone damage are important clinical outcomes in the chronic process of RA. In order to investigate whether serum CK levels could predict joint damage, we correlated CK levels at baseline and during the follow-up period with modified Sharp scores and progression. In the literature, female gender and rheumatoid factor are known for their positive relation with progression of joint damage in RA36 and might therefore act as confounding factors. However, the levels of CCL18 and CXCL16 were not related to gender or rheumatoid factor in our study population (data not shown). In these patients, neither CCL18 nor CXCL16 correlated significantly with Sharp score at baseline (lower panel of table 2). For comparison, the correlation of baseline DAS28 with progression in Sharp score between 0–3 years and 0–6 years was R = 0.36 (p = 0.005) and R = 0.39 (p = 0.002), respectively. Also, the CK levels over time, averaged over 3 years and 6 years, did not correlate with progression of joint damage over the same time period, in contrast to the DAS28 (lower panel of table 3).
CCL18 decreases upon anti-TNF-α treatment and correlates with DAS-28
Anti-TNF-α treatment is known for its strong and rapid effects on disease activity in RA. Furthermore, TNF-α has been suggested to play a role in the regulation of expression of both CCL1810 and CXCL16.9 17 To investigate the relation between chemokine levels and disease activity during treatment with anti-TNF-α, we measured these parameters in a group of RA patients that were treated with infliximab (n = 44). Interestingly, in 95% of the patients (35/37, for 7 patients, week 2 serum was not available), CCL18 levels dropped significantly (p<0.0001) after the initiation of anti-TNF-α treatment (fig 2a). In this period, the change in CCL18 was positively correlated with the change in DAS-28 (R = +0.38 (p = 0.04)). CXCL16 levels were also significantly lower (p = 0.04) at week 2, but the difference was small (median 4118 pg/ml vs. 3928 pg/ml). Moreover, CXCL16 dropped in only 65% (24/37) patients after initiation of treatment and increased in 13 patients (fig 2b), and a correlation with DAS-28 did not reach statistical significance (R = +0.38 (p = 0.07)). The CCL18 changes over the first 2 weeks of treatment did not evolve in a common pattern that could be observed throughout the whole follow-up. After 14 weeks of treatment, CCL18 and CXCL16 levels were still significantly lower than baseline CCL18 levels (fig 2c,d). However, the change in CCL18 after 14 weeks did not significantly correlate with a change in DAS-28 (R = 0.15 (p = 0.5)). As for CCL18, CXCL16 levels at week 14 did also not correlate significantly with DAS-28 (R = −0.03 p = 0.8).

{kind=link}
{kind=link}
DISCUSSION
In the present study, we show that circulating CCL18 but not CXCL16 levels are elevated in RA patients compared with controls and correlate with disease activity. In addition, neither CXCL16 nor CCL18 correlated with the level of joint damage or, more importantly, the progression of such damage.
The correlation between CCL18 and disease activity might reflect a role for CCL18 in the pathogenesis of RA. The interpretation of this observation, however, is difficult and hampered by the lack of knowledge on the exact role of CCL18 in the immune system, which might be either pro- or anti- inflammatory. It is tempting to speculate that CCL18 acts as an anti-inflammatory mediator in RA, since its production is regulated by IL-4, IL-13 and IL-10,11 25 which places CCL18 in a Th2 or regulatory corner. This would imply that serum CCL18 levels may lag behind disease activity in time. However, these thoughts are hypothetical and have not been proved so far in experimental settings. On the other hand, CCL18 might also elicit a pro-inflammatory response in RA, since the inflammatory environment might direct newly attracted T cells by CCL18 into an undesirable state of activation, subsequently resulting in ongoing T cell activation. When this hypothesis is true, CCL18 might play an active role in the chronic phase of RA. In that case, serum CCL18 will reflect disease activity more directly.
Little is known on the kinetics and dynamics of CCL18 in vivo. Since, in our inception cohort, both CCL18 levels and clinical data were measured every 3 months, this relatively long time between two measurements might influence the strength of the correlation between CCL18 and DAS-28 over time. The fact that we observed the strongest correlation within the first 2 weeks after treatment initiation in the anti-TNF-α cohort may support this. However, the decrease in correlation strength over time may also be caused by the initiation of a new treatment regimen. Effects of DMARDs on immune cells are poorly understood but may result in altered secretion of cytokines and/or chemokines. As a result, this may influence the reflection of the disease course by soluble mediators such as CCL18, which might explain the decrease in correlation strength over time. Next to the time between observations, another possible confounding factor is that elevated CCL18 levels may reflect not only disease activity in the joints but also organ involvement or comorbidity. In systemic sclerosis, for instance, a first indication for a correlation between CCL18 and pulmonary fibrosis has recently been described.38 Pulmonary fibrosis is a clinical feature that is also known to occur in RA and therefore might be an additional source of CCL18 levels in certain patients. However, of the patients included, only five patients had pulmonary comorbidity, with no case of recorded pulmonary fibrosis (data not shown).
The role of TNF-α in the regulation of CCL18 is still unclear. TNF-α blockade decreased CCL18 mRNA expression in vitro when administered to DC cultures during maturation, but administration of TNF-α to monocytes did not result in an enhanced secretion of CCL18.11 After an initial decrease in CCL18 levels in 95% of the patients, accompanied by a clear correlation with DAS-28, we did not observe any significant correlation between changes in CCL18 and DAS-28 after 14 weeks in our anti-TNF-α cohort. One could speculate that the start of anti-TNF-α treatment initially results in a decrease in CCL18 levels due to a direct effect of TNF-α blockade. In a later stage, the immune system has to search for a new equilibrium without the presence of TNF-α, which could explain why CCL18 levels do not remain low in all patients. The effects of these changes in the immune system may overrule the effect of the changes in disease activity on CCL18 levels. Thus, CCL18 levels might also be affected by the type of treatment, independently of disease activity.
We and others demonstrated that CXCL16 levels are particularly high in RA synovial fluid,9 39 which is in concordance with the high expression of CXCL16 in RA synovial tissue.9 39 CXCL16 levels were not elevated in RA serum at baseline compared with healthy controls, and we did not find a significant correlation with clinical disease parameters. This suggests that serum CXCL16 is not useful as a clinical marker in RA. However, this does not imply that CXCL16 does not play a role in the pathogenesis of RA. In the first place, soluble CXCL16 does not represent the total CXCL16 expression, since another significant portion is still membrane-bound. In order to draw conclusions with regard to a role for CXCL16 in the pathogenesis, membrane bound CXCL16 and the levels of proteases such as ADAM-10 should be taken into account also. Second, the data that are currently available on the role of CXCL16 and its receptor CXCR6 in RA point towards a role in local synovial inflammation and are not suggestive for a role in systemic inflammation. Circulating levels of inflammatory mediators do not necessarily reflect local expression in the tissues. For example, circulating levels of TNF-α hardly point towards a significant role in RA, which in fact is well appreciated in daily clinical practice. Given the data from the present study, it appears that circulating CXCL16 levels, not being elevated in RA and with no correlation with clinical parameters, do not reflect high local levels in the joints. With regard to the relation between TNF-α and CXCL-16, it is interesting that CXCL16 levels decrease significantly upon treatment with anti-TNF-α, without any significant correlation with disease activity, which was different from our previous observations in the synovial architecture.9 Whether the decrease in circulating CXCL16 levels upon neutralisation of TNF-α is a direct or indirect effect remains to be elucidated. The exact regulation of CXCL16, which may provide more insights in the role of CXCL16 in RA and may help to explain this finding is currently under our investigation.
In summary, we show that elevated CCL18 levels correlate positively with disease activity but not joint damage in RA. This correlation may reflect a role in RA pathogenesis, which might be pro- or anti-inflammatory. CXCL16 levels are not elevated and do not correlate with disease activity or joint damage. Both CCL18 and CXCL16 levels decreased upon treatment with anti-TNF-α, independently of disease activity. Although the strength of the correlations needs to be determined in large studies, our data do not directly support CCL18 as a novel clinical marker in RA, as correlations with disease activity were lower than those of markers as ESR and CRP. More knowledge on the role and regulation of both CCL18 and CXCL16 is needed to value their role in the pathogenesis of RA, both locally and systemically.
Acknowledgments
The authors would like to thank L. van den Bersselaar for technical assistance, C. Popa and E. Toonen for their efforts collecting clinical data and P. Welsing for his assistance with the statistical analysis of the data. We are indebted to all colleagues who participated in the generation and follow-up of our prospective RA cohort and anti-TNF-α cohort.
REFERENCES
Footnotes
Competing interests: All authors declare they have no conflicts of interest regarding this study
- Abbreviations:
- APC
- antigen-presenting cells
- CK
- chemokines
- DC
- dendritic cells
- DMARD
- disease-modifying antirheumatic drugs
- RA
- rheumatoid arthritis