Article Text

Abstract
Ankylosing spondylitis (AS) is a complex, potentially debilitating disease that is insidious in onset, progressing to radiological sacroiliitis over several years. Patients with symptomatic AS lose productivity owing to work disability and unemployment, have a substantial use of healthcare resources, and reduced quality of life. The pathogenesis of AS is poorly understood. However, immune mediated mechanisms involving human leucocyte antigen (HLA)-B27, inflammatory cellular infiltrates, cytokines (for example, tumour necrosis factor α and interleukin 10), and genetic and environmental factors are thought to have key roles. The detection of sacroiliitis by radiography, magnetic resonance imaging, or computed tomography in the presence of clinical manifestations is diagnostic for AS, although the presence of inflammatory back pain plus at least two other typical features of spondyloarthropathy (for example, enthesitis and uveitis) is highly predictive of early AS. Non-steroidal anti-inflammatory drugs (NSAIDs) effectively relieve inflammatory symptoms and are presently first line drug treatment. However, NSAID treatment has only a symptomatic effect and probably does not alter the disease course. For symptoms refractory to NSAIDs, second line treatments, including corticosteroids and various disease modifying antirheumatic drugs, are employed but are of limited benefit. Emerging biological therapies target the inflammatory processes underlying AS, and thus, may favourably alter the disease process, in addition to providing symptom relief.
- ankylosing spondylitis
- treatment
- cytokines
- spondyloarthropathy
- AS, ankylosing spondylitis
- CRP, C reactive protein
- CT, computed tomography
- DMARD, disease modifying antirheumatic drug
- ESR, erythrocyte sedimentation rate
- ESSG, European Spondylarthropathy Study Group
- GI, gastrointestinal
- HR, hazards ratio
- IBD, inflammatory bowel disease
- IL, interleukin
- MHC, major histocompatibility complex
- MRI, magnetic resonance imaging
- NSAID, non-steroidal anti-inflammatory drug
- RA, rheumatoid arthritis
- ReA, reactive arthritis
- SIJ, sacroiliac joint
- SpA, spondyloarthropathy
- TNFα, tumour necrosis factor α
- uSpA, undifferentiated spondyloarthropathy
Statistics from Altmetric.com
- AS, ankylosing spondylitis
- CRP, C reactive protein
- CT, computed tomography
- DMARD, disease modifying antirheumatic drug
- ESR, erythrocyte sedimentation rate
- ESSG, European Spondylarthropathy Study Group
- GI, gastrointestinal
- HR, hazards ratio
- IBD, inflammatory bowel disease
- IL, interleukin
- MHC, major histocompatibility complex
- MRI, magnetic resonance imaging
- NSAID, non-steroidal anti-inflammatory drug
- RA, rheumatoid arthritis
- ReA, reactive arthritis
- SIJ, sacroiliac joint
- SpA, spondyloarthropathy
- TNFα, tumour necrosis factor α
- uSpA, undifferentiated spondyloarthropathy
A nkylosing spondylitis (AS) is a complex and debilitating disease with a worldwide prevalence ranging up to 0.9%.1 Its aetiology and pathogenesis are not yet fully understood, and its diagnosis is difficult. As a result, the management and treatment of AS are often unsatisfactory. The accelerating pace of scientific and medical discovery is rapidly bridging the gaps in knowledge that impede progress towards a complete understanding, and subsequently, better management of the disease. Strategies to improve the plight of patients with AS are facilitated by comprehensive knowledge of history, pathogenesis, diagnosis, treatment, natural course, and socioeconomic impact of the disease. An overview of these aspects of AS is provided herein.
HISTORICAL PERSPECTIVE
The concept of ankylosing spondylitis
Palaeopathological studies of Egyptian mummies suggest that the disease now known as AS has afflicted humankind since antiquity.2,3 However, what may be the first historical description of AS did not appear in the literature until 1559 when Realdo Colombo provided an anatomical description of two skeletons with abnormalities typical of AS in his book De Re Anatomica.4 In 1693, more than 100 years after Colombo’s description, Bernard Connor, an Irish doctor, described a disinterred human skeleton that had a spine with a marked curvature. Additionally, the ilium, sacrum, five lumbar and 10 thoracic vertebrae, and five right and three left ribs appeared to be fused at “the joinings,” resulting in one continuous bone. Connor subsequently described the possible consequences of spinal curvature on movement and respiration during the patient’s lifetime.5–7
Other clinical descriptions of conditions resembling AS did not appear again in the literature until the mid-1800s. Several doctors (Lyons, Adams, Todd, Hare, Brodie, Wilson, Brodhurst, Hilton, Von Thaden, Fagge, and Sturge) reported this condition between 1831 and 1879.8 However, the reports of Wladimir von Bechterew in Russia (1893),9 Adolph Strümpell in Germany (1897),10 Pierre Marie in France (1898),11 and Connor5 are variously cited as the first descriptions of AS. Von Bechterew’s classic description of AS gave rise to the term Bechterew’s disease, used most commonly in Germany. Although these early anatomical and clinical descriptions established AS as a discrete disease entity, the concept of AS evolved with the emergence of roentgenology and other advances in science and medicine.
Roentgenology was discovered by the German physicist, Wilhelm Conrad Roentgen, at about the same time as Strümpell and Marie described AS (in 1895) but was not applied to the diagnosis or treatment of AS until the early 1920s. Roentgenographic manifestations of AS, including sacroiliitis in early disease and syndesmophytes in advanced disease, were described by Krebs, Scott, Forestier, and Robert in the 1930s.12,13 These descriptions helped to elucidate the clinical course of AS and are still applied today in the diagnosis and staging of the disease.
By the mid-1900s, radiographic, epidemiological, and clinical reports disclosed relationships between AS and several other forms of arthritis, including Reiter’s disease, psoriatic arthritis, AS, and arthropathies associated with intestinal disease.12–14 As a result, the concept of the spondyloarthropathies (SpAs) was introduced by Moll et al as a family of interrelated disorders sharing clinical and genetic characteristics distinct from rheumatoid arthritis (RA).14 The original group of disorders known as SpAs included AS, Reiter’s syndrome (reactive arthritis (ReA)), psoriatic arthritis, juvenile onset SpA (a subgroup of juvenile chronic arthritis), and arthritis associated with inflammatory bowel disease (IBD).14–16 In 1991, the European Spondylarthropathy Study Group (ESSG) modified this disease grouping to accommodate undifferentiated forms of SpA (uSpAs).17
Among the many landmarks in the history of AS and its relationship to the other SpAs, perhaps the most important were the revelations of an infectious aetiology and a genetic predisposition to AS. With respect to the latter, medical historians consider the discovery of the human leucocyte antigens (HLAs) in the 1940–50s and the subsequent characterisation of the human major histocompatibility complex (MHC) as the most important contribution to the understanding of the SpAs.8 An infectious aetiology was originally proposed based on the correlation between AS and Reiter’s syndrome, perhaps the best understood of the SpAs. In 1916, Reiter’s syndrome was described by Hans Reiter as non-gonococcal urethritis, peripheral arthritis, and conjunctivitis following dysentery.18 Subsequent documentation of the syndrome following dysentery, Shigella flexneri infection, and venereally acquired genitourinary infections established the relationship between Reiter’s syndrome and preceding gastrointestinal or genitourinary infection.19,20 The term “reactive arthritis” was introduced in 1969.21 The presence of some of the clinical signs of AS (for example, spondylitis and uveitis) in patients with ReA suggested a correlation between the two diseases. This hypothesis was confirmed in 1973 by the discovery of a high frequency of HLA-B27 in both AS and Reiter’s syndrome.8,22,23 Based upon its clinical and genetic association with ReA, it was suggested that AS also had an infectious pathogenesis. Indeed, enteric infections with Klebsiella pneumoniae and Escherichia coli have been implicated in the pathogenesis of AS in genetically susceptible hosts.24,25 Furthermore, observation of a close link between IBD and AS suggested that normal gut bacteria might stimulate the immune system once the mucosal barrier was broken.26
Landmarks in treatment
For several decades, beginning in the 1920s, x ray treatment was used to treat the spinal pain of AS with good results, including short term subjective improvement, enabling the reduced use of antirheumatic and analgesic drugs.12,27 However, x ray treatment was abandoned because of serious long term side effects, notably bone marrow effects resulting in increased risk of mortality from leukaemia and other haematological cancers, and increased risk of other malignancies.28–31 Furthermore, radiation therapy had no effect on the progression of AS.27
The analgesic properties of salicylates and opiates led them to be among the first treatments offered to patients with AS. Salicin, contained in willow and poplar barks, has been used since ancient times to treat pain, gout, and fever.32 In 1838, salicylic acid was isolated from salicin, and was noted to be an effective analgesic and antipyretic agent for the treatment of “acute and chronic rheumatism.” Aspirin, a salicylate with reduced toxicity, was developed in 1899. Although aspirin is effective in RA, it offers no therapeutic benefit in AS.33 Phenylbutazone was subsequently introduced into clinical practice in 1949 and became the first drug for which the term non-steroidal anti-inflammatory drug (NSAID) was applied. Although it was highly effective in controlling pain and inflammation,34–36 phenylbutazone was found to cause serious side effects, notably aplastic anaemia and hepatic injury, which were sometimes fatal.37,38 Thus, phenylbutazone was replaced by newer NSAIDs with improved safety profiles as first line drug treatment in AS.
In 1965, a second generation of NSAIDs led by indometacin, which demonstrated a high degree of efficacy in AS, began to be used.39–42 Many other NSAIDs soon followed, including ibuprofen, fenoprofen, ketoprofen, flurbiprofen, naproxen, diclofenac, tolmetin, piroxicam, tenoxicam, nabumetone, diflunisal, and sulindac. Results of comparative studies demonstrated that these second generation NSAIDs were equally effective in AS41–47 and were generally as effective as phenylbutazone.34–36,48 Equivalency with phenylbutazone is represented by the results of a double blind, randomised, six week comparative study of 27 patients with active AS, which found flurbiprofen (150–200 mg/day) to be as effective as phenylbutazone (300–400 mg/day) in relieving pain and tenderness of affected joints.34 Both treatments also produced significant improvement in end point parameters of spinal motion (except in the Schöber test in the flurbiprofen group and chest expansion in the phenylbutazone group). Similarly, a 12 week, randomised, double blind trial that included a 36 week open extension phase demonstrated that diflunisal (1000 mg/day) and phenylbutazone (400 mg/day) were equally effective in providing sustained relief of AS symptoms.36 Whereas diflunisal provided a more rapid onset of analgesia, phenylbutazone produced a greater increase in spinal mobility. Results of two double blind, randomised studies are representative of the comparability of the newer NSAIDs with indometacin.41,43 The first study involving 26 patients with active AS showed that flurbiprofen (150–200 mg/day) was as effective as indometacin (75–100 mg/day) in relieving pain and tenderness of affected joints after six weeks of treatment in patients with active AS.41 Patients’ and investigators’ overall subjective assessments showed improvement in 90% of patients treated with flurbiprofen and in 75% of those treated with indometacin. Significant improvements were seen in the Schöber test for evaluation of lumbar spine range of motion in the flurbiprofen group, and for chest pain in the indometacin group. The second study demonstrated equivalent efficacy between diclofenac (75, 100, or 125 mg/day) and indometacin (75, 100, or 125 mg/day) in improving all efficacy variables in patients with AS after 13 weeks of double blind treatment.42
The results of these studies, however, should not imply that all patients respond equally to all NSAIDs; individual variations in response to a given NSAID may occur. Thus, successive trials of different NSAIDs may be required.33 Although these drugs possess anti-inflammatory properties, they also frequently cause adverse gastrointestinal (GI) effects. The newest generation of NSAIDs that selectively inhibit cyclo-oxygenase-2 is associated with reduced risk of serious GI complications,49,50 but does not provide efficacy greater than the older NSAIDs.50 NSAIDs remain the preferred drug treatment for AS.33
The anti-inflammatory properties of corticosteroids were demonstrated several years before the introduction of phenylbutazone. Corticosteroids and various disease modifying antirheumatic drugs (DMARDs) were and are used for patients refractory to, or intolerant of, NSAID treatment. Although it is the general feeling among rheumatologists that corticosteroids are much less effective in SpAs than in other rheumatic diseases such as RA, unfortunately, no study is available to confirm this belief. Furthermore, corticosteroids are associated with numerous side effects, especially when given systemically over long periods of time. Direct injection of a corticosteroid into an affected joint guided by magnetic resonance imaging (MRI), computed tomography (CT), or fluoroscopy appears to be the most effective route of administration.51–54 Intravenous corticosteroid pulse therapy has also been shown to provide temporary relief of painful acute attacks.53
The DMARDs introduced between the 1930s and the 1990s, including gold salts (1930s), antimalarial drugs (1950s), d-penicillamine (1960s), sulfasalazine (1970s), and various immunosuppressive treatments (1970s to the 1990s), have not been shown to be as effective in AS as they have been in RA.33 There have been mixed results with sulfasalazine and one of its moieties, mesalazine.55–57 Methotrexate, the most commonly prescribed DMARD for RA, was reported to be beneficial in AS in open studies,58,59 but this could not be confirmed in a recent NSAID controlled, 12 month trial.60 As a result, no effective disease modifying treatment has been established for AS.
Biological agents are emerging as drugs that for the first time may provide more than just symptomatic relief to patients with AS. The anti-tumour necrosis factor α (TNFα) therapies, infliximab and etanercept, target the specific inflammatory processes of the disease, and thus, may potentially influence disease progression.
Impact of new science on ankylosing spondylitis
The completion of the human genome mapping and advances in immunology have enabled researchers to rapidly increase their knowledge of the aetiology and pathogenesis of AS and to create better treatments. The genetic basis of SpA is evident in the familial aggregation of the diseases and the strong associations with HLA-B27.8
Presently, at least 23 subtypes of HLA-B27 have been identified (B*2701 to B*2723), although they are not equally distributed throughout the world.61,62 The only differences between alleles associated with disease and those that are not amount to two amino acid residues found at the bottom of the peptide binding groove of HLA-B27.62 These sites may prove to be a key in characterising the pathogenesis of SpA.
Occurrence of AS or related SpA has been documented in patients possessing any one of the first 10 subtypes.61 However, it appears that the subtypes, B*2706 and B*2709, may not be associated with, or have a limited association with, AS. For example, most native Indonesians have subtype B*2706, but SpA is rarely seen in this population. None the less, it must be noted that patients carrying alleles such as B*2706 are known who have SpA.63 Similarly, B*2709, which is frequent in Sardinia, does not seem to be associated with AS.61 This subtype differs only in one amino acid at the bottom of the peptide binding groove. These findings suggest that one pathogenic peptide, presented by all other B27 peptides but not by B*2706 and B*2709, may have a central role in the pathogenesis. To date, such a peptide has not been identified.
The HLA-B27 gene has been cloned, sequenced,64,65 and introduced into rats.66 Rats from one of these transgenic lines expressing HLA-B27 spontaneously developed an inflammatory syndrome closely resembling the HLA-B27 associated human disorders. The transgenic model might therefore be a valuable tool for studying AS pathogenesis.
CLASSIFICATION OF THE SPONDYLOARTHROPATHIES
Spondyloarthropathies as a group
The SpAs are defined as inflammatory arthropathies characterised by sacroiliac involvement and relationship to HLA-B27. They are differentiated from RA by distinct clinical features, association with HLA-B27, and an overlap between the individual SpA diseases.26 The true spectrum of SpA ranges beyond the originally defined group of disorders.14–16 Owing to lack of adequate criteria, uSpAs, including seronegative oligoarthritis, dactylitis or polyarthritis of the lower extremities, heel pain due to enthesitis, and early sacroiliitis without radiologically detectable changes, were overlooked. Thus, in 1991, the ESSG developed criteria to incorporate the uSpAs (table 1).17,67 The current subcategories of SpA are AS, ReA, psoriatic arthritis, IBD associated arthritis, and uSpA.68
Classification of spondyloarthropathies using ESSG criteria17
Ankylosing spondylitis
AS is the prototype of the SpAs and one of the common rheumatic diseases.1 Sacroiliitis is the earliest recognised manifestation of AS, but peripheral joints and extra-articular structures may also be affected. Subchondral tissues become granulomatous and infiltrated with plasma cells, lymphocytes, mast cells, macrophages, and chondrocytes. The affected joints show irregular erosion and sclerosis. Tissue is gradually replaced by fibrocartilage and then becomes ossified. When these lesions occur in the spine, the junction of the annulus fibrosus of the disc cartilage and the margin of the vertebral bone undergo irreversible damage. The outer annular fibres are replaced by bone and the vertebrae become fused. In advanced stages of the disease the fusion typically ascends the spine, forming a long bony column referred to as “bamboo spine.”
The only clinical sign currently used to differentiate AS from sacroiliitis present in patients with uSpAs is radiographic evidence of ≥grade II bilateral or >grade III unilateral sacroiliitis. As seen in a 10 year follow up study of 88 patients with possible AS, the prolonged course of the disease delays differentiation of AS from uSpA.69 This study found the progression from uSpA to definite AS, as shown by radiological sacroiliitis, to occur after at least 9±6 years; radiological signs of spinal involvement were apparent much later (after 11±6 years of disease duration) (fig 1).

Evolution from undifferentiated spondyloarthropathy (uSpA) to ankylosing spondylitis (AS) in patients with definitive radiological sacroiliitis.69 This previously unpublished figure is based on data published by Dr W Mau and is printed with the author’s permission.
EPIDEMIOLOGY
Age of onset
AS commonly starts in the second or third decade of life.70,71 A survey of 3000 German patients with AS showed the following distribution pattern of age at the time of first spondylitic symptoms: 4% were younger than 15 years; 90% were 15–40 years; the remaining 6% were more than 40 years.72 Analysis of a German rheumatological database (n=8776) determined a mean age at onset of AS of 28.3 years.73 The clinical picture of early (juvenile onset) AS differs from that of adult onset by the more frequent involvement of peripheral joints.70 A cohort study dividing patients according to age of symptom onset found (a) a higher prevalence of hip involvement among patients with young age at onset, and (b) a striking increase in the prevalence of total hip replacements in those with juvenile onset AS (18% compared with 8% for adult onset, p<0.001).73 The difficulty in diagnosing AS in its early stages is evident in the difference between age at onset reported above and age at diagnosis (mean (SD) age 32.7 (8.6) years).74 Juvenile onset SpA is explained in more detail in the article “Juvenile onset spondyloarthropathies” within this supplement (p iii33).
Sex
Men are afflicted with AS approximately two to three times more frequently than women.71 Estimated percentages of male patients among the AS patient population range from 65% to 80% and vary by geographic location (68.9% in a German rheumatological database, n=877671; and 78.3% in a French study, n=473).75 The disease pattern varies by sex.76–78 The spine and pelvis are most commonly affected in men, with some involvement of the chest wall, hips, shoulders, and feet. In contrast, women have less severe involvement of the spine, with more symptoms in the knees, wrists, ankles, hips, and pelvis.76–78 Disease also tends to be more severe in men.77
Prevalence
For the SpAs as a group, the overall prevalence in the population has been reported to be as high as 1.9%.1 There is a wide geographic variation in reported estimates of the prevalence of AS. However, in general, there is a close correlation between the prevalence of HLA-B27 and the prevalence of SpAs in a given population. Among the total 3.47 million population of Berlin, Germany, the prevalence of AS estimated from an HLA-B27 frequency of 9.3% was reported to be 0.86%.1 The reported adult prevalence of AS in Finland was 0.15%,79 and 1.1–1.4% (men 1.9–2.2%, women 0.3–0.6%) among adults in Norway.80 The overall prevalence of SpA among adult Eskimo populations in two study regions in Alaska was estimated at 2.5%.81 Prevalence also appears to vary among ethnic groups. The estimated nationwide prevalence of SpA among the total Japanese population (9.5/100 000) is less than 1/200 of that among white subjects.82
PATHOGENESIS
The pathogenesis of AS is poorly understood. Immune mediated mechanisms are suggested by inflammatory histology, raised serum levels of IgA and acute phase reactants, and the close relationship between HLA-B27 and AS. No single agent or event has been identified as the cause of the disease, but the interrelationship between AS, ReA, and IBD suggests that enteric bacteria may play a part.8,83
Histopathology
Enthesitis, defined as “the inflammatory changes of an enthesis”,84 is considered a characteristic finding in AS and other SpAs. Enthesis refers to the insertion of a tendon, ligament, capsule, or fascia into bone.84,85 The enthesis encompasses the inserted structure and the bone to which it is attached.84 The pathological changes of enthesitis, especially in the early stages, have been difficult to study for technical and ethical reasons. The importance of enthesitis relative to synovitis, subchondral marrow inflammation, and osteitis in AS is under debate.86 However, more recent work suggests that the entheseal fibrocartilage is the major target of the immune response and the primary site of the immunopathology. Theoretically, immunocompetent cells could get access to fibrocartilage derived antigens from bone marrow derived blood vessels.87 It has been suggested that not only fibrocartilage but also cartilage in general at the interphase with bone should be regarded as the primary site.88
Enthesitis was originally considered as the hallmark of AS on the basis of findings from two cases of advanced AS in the 1970s.89 A more recent study evaluated changes in the sacroiliac joints (SIJs) of 12 patients with AS, including five biopsies, and compared them with 22 control necropsy cases.90 Mild but destructive synovitis and myxoid subchondral bone marrow were the earliest changes identified in SIJs from patients with AS. The adjacent articular tissues were destroyed by these lesions and were followed by varying degrees of fibrous scarring, woven bone, and new cartilage formation. Both original and new cartilages were replaced by bone through fusion; the predominant mode of ankylosis was chondral fusion.90,91
The first immunohistological examination of entheses was performed in samples taken from eight patients with SpA and compared with those from patients with RA (n=4), or osteoarthrosis (n=3).92 Enthesis samples of the vastus lateralis muscle or of the cruciate ligament were taken during joint replacement. The bone marrow of the SpA samples showed oedema and contained cellular infiltrates. The density of all cell types in the bone marrow was significantly higher in patients with SpA than in patients in the two other groups. There were also more CD8+, CD3+, CD4+, and CD20+ cells in the SpA group. In particular, the CD3+ cell subset was increased fivefold in the SpA group compared with the RA group. Within the SpA group, the predominant T cells were CD8+ cells.
Pathological studies have shown that inflammatory infiltration and destruction are not restricted to the enthesis of the intervertebral disc, but rather affect the whole annulus fibrosus, which also consists of fibrocartilage.93 In the recent past, MRI studies have helped considerably to define the primary site affected in SpA. These studies show primarily an osteitis with bone marrow oedema at the cartilage/bone interphase, which correlates nicely in the acute phase with the infiltration of mononuclear cells.94 These cells, most probably T cells coming from the bone marrow, invade the cartilage (fig 2).90,94 This targeted, probably cartilage derived, antigen has not yet been identified.

A biopsy specimen from a patient with acute sacroiliitis shows cellular infiltrate containing activated fibroblasts and lymphocytes that seem to invade a degenerate cartilaginous area.94 Reproduced with permission of the author and the copyright holders from reference 94. Copyright © 2000 by the Annals of the Rheumatic Diseases.
Genetic, immunological, and environmental factors
Some authors believe that the interaction between the class I MHC molecule HLA-B27 and the T cell response is a key to the pathogenesis of AS. A pathogenic antigen presented by HLA-B27 to CD8+ T cells could be derived from fibrocartilage/cartilage, as discussed above.95 Based upon studies of siblings and twins, which suggest only 16–50% of total genetic risk for disease,96,97 it is believed that genes outside the HLA region must be involved.98 The non-concurrent development of AS in twins, especially monozygotic twins (concordance rate of about 75%), suggests that environmental factors also may play a part in pathogenesis. Although no other genes have been proved to be responsible for AS, potential candidates include certain MHC class I and MHC class II genes, and non-MHC genes.98,99 Genome-wide screens have identified other susceptibility regions on chromosomes 1p, 2q, 6p, 9q, 10q, 16q, and 19q.100
Bacterial infections are suggested to be triggering events in the pathogenesis of SpA, as documented in ReA.101 Because bacterial DNA, RNA, and proteins can be detected in ReA affected joints, it is believed that the subsequent immune response triggers the arthritis.102 T cells with specificity for these bacteria have been isolated in synovial fluid and peripheral blood of patients with ReA.103,104 The close relationship between AS and inflammation of the gut mucosa, associated with clinical or subclinical forms of IBD, suggests that normal gut bacteria and, subsequently, immune reaction directed against gut bacteria, may also participate in the pathogenesis of AS.105
There is evidence that the pattern of cytokine secretion influences the pathogenesis of SpA.101 The percentage of T cells secreting TNFα and interferon γ has been found to be lower in the peripheral blood of patients with AS and healthy HLA-B27 positive control patients than in HLA-B27 negative control patients.106 Patients with AS were found to have a higher production of IL10 by CD8+ T cells compared with either of the control groups. Low TNFα and interferon γ, and high IL10 levels, also occur in ReA.107,108 Several studies reviewed by Rudwaleit and Hohler101 suggest that changes in production of TNFα and IL10 may be partially determined by genetic polymorphisms. A relative deficiency of T helper (Th)1 cytokines such as TNFα might lead to longer persistence of bacterial antigens at the beginning of the immune response. Such prolonged antibacterial immune responses could then trigger an autoimmune response.95
Studies attempting to explain the sex bias in AS have shown no evidence of a sex linked genetic factor109 or a hormonal (androgen) factor.110 In a linkage study of the X chromosome of 234 sibling pairs affected by AS, Hoyle et al found no correlation between the X chromosome and susceptibility to AS.109 Mori et al found no conclusive evidence of an association between AS and the androgen receptor gene activity.110
DIAGNOSIS
The diagnosis of AS before the occurrence of irreversible damage is difficult. Several years may pass between onset of symptoms and definite diagnosis. This delay is most likely due to low awareness among non-rheumatologists of AS or SpA and the fact that radiological proof of sacroiliitis is a late feature of the disease.69,72 This is unfortunate, as earlier diagnoses might potentially reduce the crippling effects that can occur.
Risk factors
The risk factors that predispose a person to AS include (a) HLA-B27 seropositivity; (b) family history of AS; (c) male sex; and (d) frequent GI infections.111 A comparison of relatives of patients with AS and the general population determined that the risk for AS was 16 times greater among HLA-B27 positive relatives (21% had AS) than among HLA-B27 positive individuals from the general population (1.3% had AS).112 The HLA-B27 negative relatives did not have any manifestations of AS. As discussed earlier, AS occurs more commonly in men than women. The deficiency in TNFα secretion by T cells, coupled with the increased levels of IL10 as seen in ReA,108 also may result in long term persistence of bacteria, leading to inflammation and subsequent pathogenesis in AS.113,114
Clinical manifestations
Symptoms
The first symptoms of AS usually appear in late adolescence or early adulthood. The initial symptom is typically a dull pain that is insidious in onset. The pain is generally felt deep in the buttock and/or in the lower lumbar regions and is accompanied by morning stiffness in the same area that lasts for a few hours, improves with activity, and returns with inactivity. The pain becomes persistent and bilateral within a few months and is usually worse at night. About 5% of patients presenting with chronic inflammatory back pain have AS or another SpA subset.115 The prognostic importance of inflammatory back pain lies in the likelihood of future progression to definite AS.69
For some patients, bone tenderness may be the primary complaint or may accompany back pain or stiffness. Arthritis in the hips and shoulders occurs in some patients, often early in the course of the disease. Asymmetric arthritis of other joints, predominantly of the lower limbs, can be present at any stage of the disease. Neck pain and stiffness is characteristic of advanced disease.
There are several extra-articular manifestations of AS, the most common condition being acute anterior uveitis. Patients may present with unilateral pain, photophobia, and increased lachrymation. Up to 60% of patients with AS have asymptomatic IBD.116,117 In some cases, frank IBD will develop.117 Aortic insufficiency, with possible congestive heart failure, is seen infrequently in patients with AS.
Physical findings
A principal physical finding is loss of spinal mobility, with restrictions of flexion, extension of the lumbar spine, and expansion of the chest. The limitation of motion is disproportionate to the degree of ankylosis because of secondary muscle spasms. Pain in the SIJs may be elicited with direct pressure or movement, but its presence is not a reliable indicator of sacroiliitis. There may be detectable inflammation of peripheral joints. Clinical signs of the disease can range from mild stiffness to a totally fused spine, with any combination of severe bilateral hip involvement, peripheral arthritis, or extra-articular manifestations. A patient’s posture undergoes characteristic changes if a severe case goes untreated. The lumbar lordosis is destroyed, the buttocks atrophy, the thoracic kyphosis is exaggerated, and the neck may stoop forward.
Laboratory findings
Although no laboratory test is diagnostic of AS, the HLA-B27 gene is present in about 90–95% of white patients with AS in central Europe and North America.1 Only 50–70% of patients with active disease will have an increased level of C reactive protein (CRP) and a raised erythrocyte sedimentation rate (ESR).69,118–120 However, measurement of the levels of these acute phase reactants appears to have limited value in determining disease activity.118,121,122 Studies have shown a lack of correlation between clinical signs of disease activity (pain, stiffness, and sleep disturbance) and CRP and ESR.118,122 Mild normochromic normocytic anaemia may be detected. A raised alkaline phosphatase level may be present in severe disease. Above normal serum IgA levels are common. Synovial fluid from affected limbs does not differ in appearance from that of any inflammatory joint disease. Airflow measurements and ventilatory function remain normal in patients with restricted chest wall motion, but vital capacity is decreased and functional residual capacity is increased.
Radiographic findings
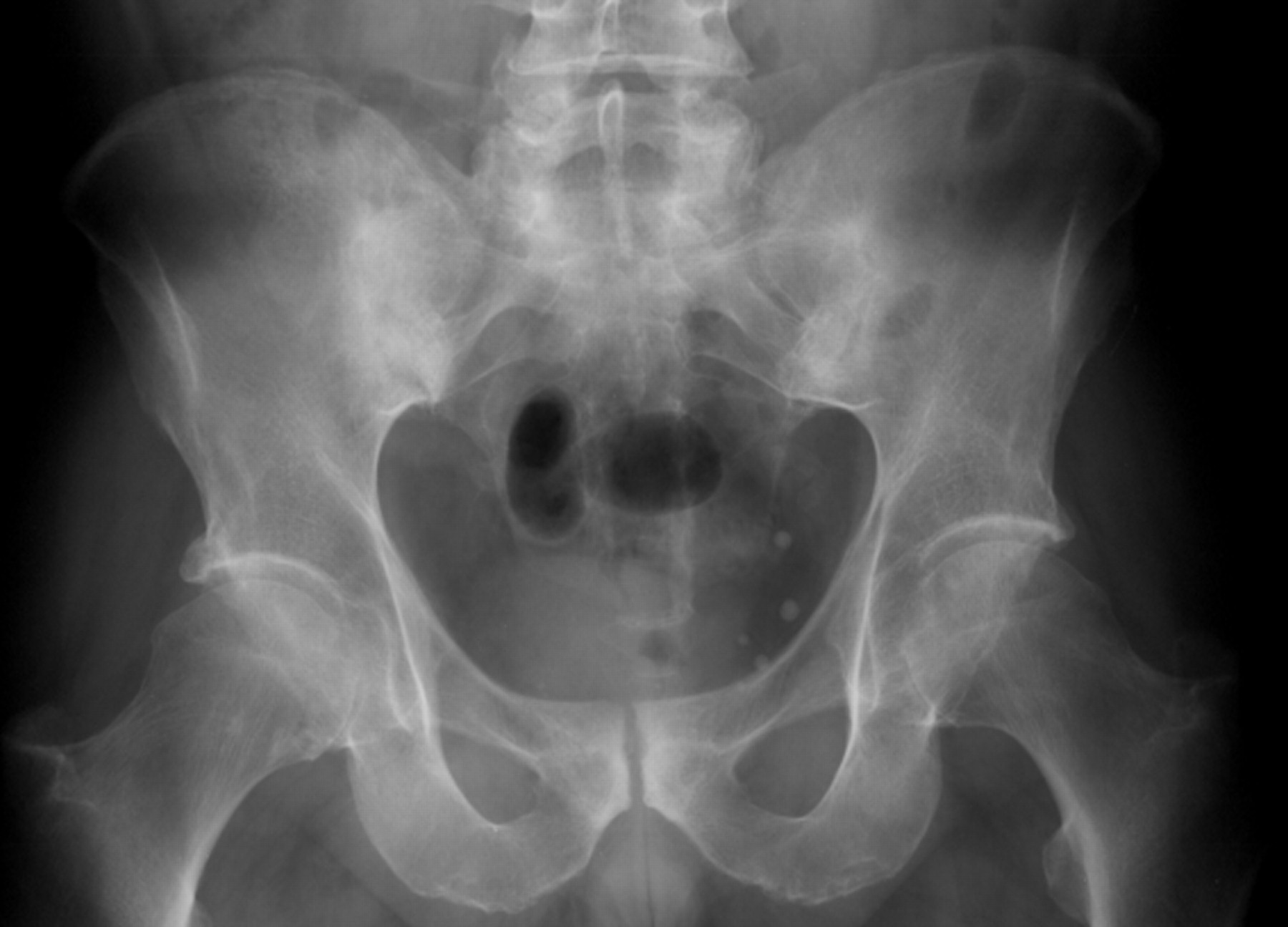
Radiological changes reflect the disease process; thus, radiographic sacroiliitis usually becomes apparent at some point during the course of AS. However, many years of disease may pass before unequivocal sacroiliac changes are evident on radiographs. The earliest visible changes in the SIJs are blurring of the cortical margins of the subchondral bone, erosions, and sclerosis. As erosion progresses, the joint space appears wider, and then fibrous and bony ankylosis obliterates the joint. Joint changes usually become symmetric during the course of the disease. The New York grading system for sacroiliac joint status is as follows: grade I=suspicious; grade II=evidence of erosion and sclerosis; grade III=erosions, sclerosis, and early ankylosis; and grade IV=total ankylosis.123 Figure 3 shows radiographs of a patient with AS displaying grade II and grade II–III sacroiliitis. Figure 4 shows a radiograph of a patient with AS displaying grade III sacroiliitis.

Pelvic radiograph of a patient with AS showing sacroiliitis grade II on the right side and grade II–III on the left. This previously unpublished figure is provided courtesy of Dr M Rudwaleit.

Pelvic radiograph of a patient with AS showing bilateral sacroiliitis grade III. This previously unpublished figure is provided courtesy of Dr M Rudwaleit.
CT and MRI can detect AS lesions earlier and with greater consistency than plain radiography, but these methods are not routinely employed.122,124–126 MRI, which is better than radiography for detection of early sacroiliitis, can be performed if radiographs are negative in patients with clinical signs of AS.124–126 The radiograph in fig 5A shows minimal changes, while the corresponding MRI scan (fig 5B) reveals acute inflammation. A prospective evaluation of the relative sensitivities of MRI, quantitative sacroiliac scintigraphy, and plain radiography in detecting active sacroiliitis in 44 patients with clinical symptoms of inflammatory low back pain plus additional features of SpA found MRI to be the most sensitive imaging technique (95% sensitivity, compared with 19% for plain radiography, and 48% for quantitative sacroiliac scintigraphy).126 These findings indicate that MRI enables detection of approximately 75% more cases of early sacroiliitis (AS) that would otherwise have been missed by plain radiography. CT or MRI may also be useful tools for monitoring progression of sacroiliac joint sclerosis.122 Overall, radiographic (CT and plain radiography) findings do not correlate well with disease activity.122 In one study, pain and stiffness correlated positively with an increase in sacroiliac joint sclerosis detected by CT and negatively with increasing ankylosis.122

Pelvic radiograph (A) showing suspicious changes: a small circumscribed zone of sclerosis of the left sacroiliac joint (sacroiliitis grade I–II) and slightly blurred joint margins of the right sacroiliac joint (grade I). The corresponding MRI (B) of the sacroiliac joints shows contrast enhancement of periarticular bone (oedema) and of the joint space of both sacroiliac joints reflecting acute inflammation (subtraction technique after intravenous application of gadolinium). These previously unpublished figures are provided courtesy of Dr M Rudwaleit.
Diagnostic criteria
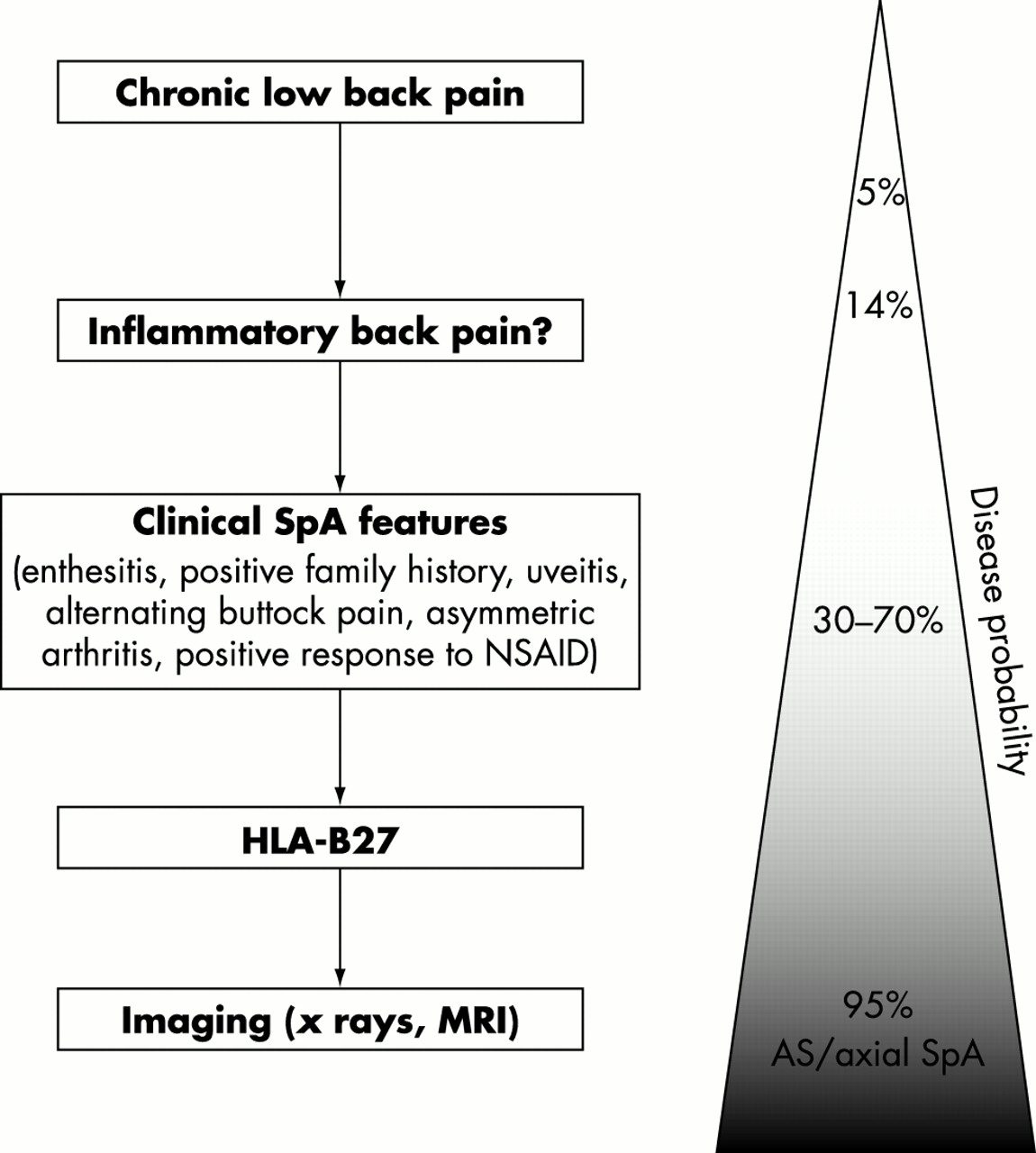
Inflammatory back pain, according to Calin et al, is present if four of the following five features are present: (a) age at onset <40 years; (b) back pain >3 months; (c) insidious onset; (d) morning stiffness; and (e) improvement with exercise.127 On the basis of the 1984 modified New York criteria,128 the diagnosis of AS can be made if radiological sacroiliitis (either grade II bilaterally, or grade III unilaterally) is present in conjunction with clinical signs (inflammatory back pain or restriction of spinal mobility) (table 2).128 However, in the absence of definite radiographic findings, one can calculate individual disease probabilities depending on the presence of typical SpA manifestations (such as inflammatory back pain, enthesitis, uveitis, asymmetric arthritis, positive family history, response to NSAIDs, HLA-B27, raised CRP). For example, the disease probability of axial SpA (early AS) in a patient with inflammatory back pain increases from 14% to around 50–60% if there are one or two more clinical SpA features present. It further increases from 50% to 90% if HLA-B27 is positive or if the MRI is positive. Thus, in patients reaching disease probabilities of 80–90%, the diagnosis of axial SpA should be made, as indicated in the diagnostic algorithm shown in fig 6. The important conclusion from the probability calculations is that an early diagnosis of axial SpA can be made with sufficient probability, even in the absence of typical radiological changes.
Modified New York Criteria for Ankylosing Spondylitis128

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Diagnostic algorithm for axial SpA (early AS) starting with the assessment of inflammatory back pain. The combination of several typical SpA manifestations results in a high disease probability. This figure will be published in Ankylosing spondylitis: clinical features. In: Hochberg M, Silman A, Smolen J, Weinblatt M, Weisman M, eds. Rheumatology. 3rd ed. London: Mosby: a division of Harcourt Health Sciences Ltd, ©2002. Reproduced with permission from Dr M A Khan and Mosby.
The value of HLA-B27 testing in the diagnosis of AS has not been clarified in the past,129 but its usefulness is strongly supported by the probability calculations. However, tests for HLA-B27 should only be carried out in a patient with inflammatory back pain and not in all cases of back pain. As indicated in fig 4, further SpA features have to be present in addition to inflammatory back pain and HLA-B27 to reach a sufficiently high disease probability to make the diagnosis.
NATURAL HISTORY
Disease course and prognosis
AS is a chronic condition with no predictable pattern of progression; thus, the disease does not follow a single defined course. Although many outcomes are possible, findings from an early prospective study suggest that a predictable pattern of AS emerges within the first 10 years of disease.130 In this study the natural course of AS was examined over a 23 year period in 51 patients; their mean disease duration was 38 years. Seventy four per cent of the patients who had mild spinal restriction after 10 years did not progress to severe spinal involvement. In contrast, 81% of the patients who had severe spinal restriction were severely restricted within the first 10 years.
Amor et al identified hip involvement or the presence of three of the following factors within two years of onset of SpA as predictive factors for severe disease (specificity 97.5%) and severe outcome (sensitivity 50%): ESR >30 mm/1st h, NSAID unresponsiveness, limitation of lumbar spine, sausage-like finger or toe, oligoarthritis, or onset at ≤16 years.131 Hip arthritis was associated with a 23-fold increase in the risk of severe disease. The absence of any of these factors during the first two years of the disease was predictive of a mild outcome (sensitivity 92.5%; specificity 78%).
A recent study found that age at symptom onset had no significant effect on radiological disease progression or disease activity.73 However, consistent with the findings of Amor et al,131 regardless of age at onset, hip involvement (arthritis) was a risk factor for radiographic spinal progression. Furthermore, hip involvement was more prevalent among patients with juvenile onset of symptomatic disease, and subsequently, total hip replacement was significantly more prevalent among patients with juvenile onset disease (18% v 8% for adult onset; p<0.001). In contrast, young onset patients without hip problems did not have more severe disease.
There appear to be some differences between the sexes in the course of the disease. Many reports show that women have a later age of onset,132 milder disease,133 and more extraspinal involvement.76 A retrospective review of 41 women and 41 men with definite AS found no statistically significant differences between the two groups in clinical presentation at onset between the two groups.77 However, the disease was less severe in women than in men as evidenced by (a) lower, albeit non-statistically significant incidence and duration of uveitis; (b) longer asymptomatic periods; and (c) significantly (p<0.05) lower leucocyte counts and lower γ globulin levels.77 At the end of the study, fewer women were unable to work because of persistent peripheral arthritis (7% v 26% of men; p<0.03) or arthrosis (17% v 39% of men; p<0.05) and more women than men remained in functional class I (75% v 46%, respectively; p<0.003). Although radiological assessment showed a significantly higher incidence of sacroiliitis among women (68%) than among men (44%; p<0.05), women had a significantly lower incidence of “bamboo spine” (12% v 34% in men; p<0.008). It should be noted that some of the patients in this study were from the Mexican mestizo population, in whom clinical expressions of AS differ somewhat from those seen in white subjects.134,135 The major difference between the two ethnic populations is higher frequency of peripheral arthritis, and fever at onset in Mexican mestizo patients.134,135
COMPLICATIONS
The most serious complication encountered in AS is spinal fracture. Even minor trauma to the rigid, fragile spinal column can cause severe damage. The cervical spine is the most susceptible site; fractures at this site can result in quadriplegia. Prostatitis is highly prevalent among men with AS. Aortic insufficiency and cardiac conduction disturbances can occur in patients with long term disease. Amyloidosis, cauda equina syndrome, and pulmonary fibrosis are rare complications.
SOCIOECONOMIC IMPACT
When considering the socioeconomic consequences of a disease, three domains need to be distinguished. Firstly, the (in-)ability to continue in paid or unpaid work, which can be valued in monetary terms as productivity costs. Secondly, the disease related health resource use, which can be valued as direct costs. Thirdly, the impact of the disease on the quality of life and psychological wellbeing, which cannot be expressed in monetary terms and which is referred to as intangible costs. Because AS starts usually at an early age, the lifetime socioeconomic impact of the disease can be important for the patient as well as for society. Until now, the socioeconomic consequences of AS have not received much attention. When assessing published reports on this subject, it is difficult to compare data from studies performed in different countries. Variations in social security and healthcare systems limit attempts at making comparisons and generalisations about socioeconomic data.
Work status and productivity costs
A recent systematic literature review including 18 articles on 14 patient groups136 and five more recently published studies reported the working status in AS.71,137–140 It was found that employment rates among patients with AS ranged from 55% to 89%, and in half of these studies, employment was below 70%. Annual days of sick leave for those with a paid job were reported in five of these studies and varied from six to 46 days for each patient.136,139,140 Work disability ranged from 3% to 41%, and in half of the studies was higher than 20%.136,137,139,140 It should be noted that most studies did not adjust data for age and sex, which might explain the seemingly favourable employment rates.
Several studies identified older age,136,138 longer disease duration,136,138 lower level of education,71,136–138 reduced physical functioning,136–138 pain,74,137,138 and more physically demanding jobs136,137 to be significant risk factors for work disability. Boonen et al examined the withdrawal from the labour force in patients who were employed at the onset of AS.74 Of 529 patients with a paid job before diagnosis, 5% had left the work force within the first year after diagnosis, 13% after five years, 21% after 10 years, 23% after 15 years, and 31% after 20 years. The age and sex adjusted risk of withdrawal from paid work was 3.1 (95% CI 2.5 to 3.7) times higher than in the general Dutch population. Within patients, determinants of withdrawal from work were older age at diagnosis, manual work, and coping strategies characterised by limiting or adapting activities. Using data from the German rheumatological database, including 52 444 patients with RA and 8776 patients with AS, Zink et al found a higher employment rate in patients with AS (71.3%, 62.5% in women, and 75.3% in men) than in patients with RA (49.5%; 45.1% in women, and 64.3% in men).71 Taking into account the different age structures and educational levels in the two diseases, the employment rates for patients with AS were still significantly higher (for example, 67.5% in men aged 51–60 with AS compared with 53.9% in RA).
Interestingly, differences were noted between East and West Germany, reflecting the higher rate of unemployment in East Germany (19.5% v 11.0% in West Germany in 1997). In the Western states, the probability of maintaining employment was 4.4% lower for men with AS and 20.2% lower for men with RA than the rate in the general population. In comparison, in the Eastern states, the employment rate was 11.4% lower for men with AS and 28.2% lower for men with RA than the rate in the general population. A recent study confirmed the influence of “country” on labour force participation. In a European three nation study, it was shown that age and sex adjusted work disability rates were higher in The Netherlands (41%) than in France (23%) or Belgium (9%) (table 3). Within each country, work disability among patients was higher than expected in the general population. When adjustments were made for differences in demographic and disease related confounders, Dutch patients with AS had a 3.82 (95% CI 1.33 to 11.0) times higher risk of being work disabled than patients living in either France or Belgium.139 It is of note that in The Netherlands and France patients can have a partial work disability while continuing in a part-time paid job, whereas such a possibility does not exist in Belgium. Annual sick leave in those with a paid job was higher in The Netherlands (19 days per patient) than in France (six days per patient) or Belgium (nine days per patient) (table 3). The difference among countries remained significant after correction for baseline sociodemographic and disease characteristics.139
Productivity costs¶ of ankylosing spondylitis in the USA, The Netherlands, France, and Belgium
Two studies calculated productivity costs associated with AS.139,141 In the longitudinal study already mentioned, among 209 patients with AS from The Netherlands, France, and Belgium, friction costs (reflecting productivity losses because of sick leave only for the average period of job vacancy) as well as human capital costs (reflecting productivity losses for the whole period of sick leave and work disability) were calculated (table 3). Average annual human capital costs were 8862 euros (median 2853), 3188 euros (median 0), and 3609 euros (median 0) per patient in The Netherlands, France, and Belgium respectively. Using Cox proportional hazard analysis, it was shown that the productivity costs were higher among patients living in The Netherlands (hazard ratio (HR) 0.63; 95% CI 0.42 to 0.96) than in both other countries, and also among those having inflammatory bowel disease (HR 0.47; 95% CI 0.23 to 0.97) as comorbidity and those with worse physical function (HR per point Bath AS Functional Index 0.89; 95% CI 0.81 to 0.97). An HR <1 indicates higher costs.139
The second study was a prospective longitudinal study conducted in the USA in 241 patients with AS.141 This study used the human capital approach to calculate productivity costs of paid and unpaid work by including the number of days of limited activity due to AS symptoms among retirees and homemakers. Average annual productivity costs per patient were $4945 (median 0). Converted to euros using 1998 purchasing parities, this would equal 4227 euros per patient per year (median 0). For comparison, the human capital cost of RA is reported to range from 924 euros a year to 15 745 euros per patient per year.142
Health resource use and direct costs
The use of healthcare resources is significant for patients with AS.141 The previously mentioned longitudinal study of 241 patients with AS in the USA estimated the average annual direct (healthcare and non-healthcare) costs generated by AS to be $1774 per patient (median $1113). Converted to euros using 1998 purchasing parities this would equal 1517 euros per patient per year (median 951 euros). Of the direct costs, the cost of drugs contributed 42% to the total, inpatient care 16%, ambulatory care 15%, private household help 12%, and technical procedures 12%. Of the total costs of the disease ($6720 per patient per year; 5744 euros per patient per year), the productivity costs represent the major portion (73.6%).142 In multivariate analysis, it was shown that functional disability was the most important predictor of total costs. For each one point increase in the Health Assessment Questionnaire disability index modified for the SpAs, the likelihood of high cumulative total costs over a five year period (>$50 000) increased by more than sixfold.
Quality of life or intangible costs
Patients with symptomatic AS have to cope with pain, sleep problems, functional disability, dependency, out of pocket expenses, and income loss. All these factors affect the patient’s general wellbeing. In a direct comparison of functional disability and pain among patients with RA and AS registered in the German rheumatological database, among men of the same age groups the rate of severe functional disability was higher in AS than in RA (at, however, much longer disease duration), whereas functional disability in women was similar in RA and AS.71 A longitudinal cross-sectional survey of 175 patients with AS identified aspects of the disease that adversely affect quality of life.143 The majority of patients surveyed were men (68%). The patients had a mean age of 51 years and had AS for a mean duration of 23.7 years. The most prevalent quality of life concerns included stiffness (90%), pain (83%), fatigue (62%), sleep problems (54%), appearance (51%), future outcome (50%), and side effects of drugs (51%). Compared with patients with some college education, patients with less education (≤12 years of education) had a significantly lower quality of life and were two to four times more likely to be concerned about drug side effects, mobility, housework, self care tasks, coping with illness, anxiety, payment for treatment, and relationships with spouses, family, and friends.
CONCLUSIONS
AS is a complex, unpredictable disease that has puzzled and frustrated clinicians and scientists alike for centuries. It is insidious in onset, striking individuals, mostly men, at an early age, subsequently progressing over several years until structural damage manifests clinically as inflammatory back pain (sacroiliitis) and loss of spinal mobility, and a definite diagnosis of AS is made. Peripheral and extra-articular symptoms may also occur. Patients with severe AS have a reduced quality of life and loss of productivity due to work disability and sick leave. In addition, the management of AS is taxing on healthcare resources. Thus, indirect and direct costs associated with AS are high.
The pathogenesis of AS is poorly understood. However, the prevailing hypothesis is that immune mediated mechanisms have a major role. Researchers are currently exploring the pathogenic role of inflammatory cellular infiltrates, including various cytokines such as TNFα, and the interaction between the T cell response, HLA-B27, and genetic and environmental factors, including bacterial antigens. The close relationship between AS and clinical and asymptomatic forms of IBD suggests the potential involvement of an immune reaction directed against gut bacteria. Sacroiliitis detected by radiography, MRI, or CT in the presence of clinical manifestations is diagnostic of AS. However, the presence of inflammatory back pain, plus at least two to three other typical features of SpA (for example, enthesitis, uveitis, HLA-B27 positivity, or raised ESR), is generally diagnostic of axial SpA, which usually progresses to AS over time. At present, NSAIDs, in conjunction with physical therapy, are the mainstay of treatment for patients with symptomatic AS. However, these measures are strictly palliative, and NSAIDs do not alter the course of the disease or prevent structural damage. For symptoms refractory to NSAIDs, second line treatments including corticosteroids and various DMARDs are employed. However, these treatments are of limited benefit. Emerging biological therapies target the inflammatory processes underlying AS, and thus, may favourably alter the disease process while providing relief of symptoms.