





Abstract
Megabase-sized, complex, repetitive regions of genomes are poorly studied, due to the technical and computational challenges inherent to both assembling precise reference sequences and accurately assessing structural variation across contiguous megabase DNA regions. Here we describe a strategy to overcome these challenges, CISMR (CRISPR-mediated isolation of specific megabase-sized regions of the genome), which enables us to perform targeted isolation of contiguous megabase-sized segments of the genome. Direct sequencing of the purified DNA segments can have >100-fold enrichment of the target region, thus enabling the exploration of both DNA sequence and structural diversity of complex genomic regions in any species.
INTRODUCTION
Throughout eukaryotic genomes there are megabase-sized regions of complex genomic structures harboring genes with important biological functions. In many cases, the reference genome sequences of these regions are incompletely assembled owing to their highly repetitive nature and huge size. Moreover, the inter-individual and inter-species variation of these regions is highly polymorphic on many size-scales (1,2), indicating a plethora of structural and functional variation is yet to be discovered. In the few cases where the DNA sequence of such regions has been accurately determined, important insights into processes such as immunity (e.g. immunoglobulin heavy-chain locus) (3) and reproduction (e.g. human Y chromosome) (4) are revealed. Nevertheless, the methods to resolve such regions are labor intensive and expensive, requiring whole genome clone libraries and haplotype-specific iterative mapping and sequencing (5). Here we describe a strategy to overcome these challenges, CISMR (CRISPR-mediated isolation of specific megabase-sized regions of the genome), which enables us to perform targeted explorations of megabase-sized segments of the genome. Recently, the specificity of CRISPR enzymology has been used to target and clone short single copy genomic sequences in vitro (6,7). CISMR extends upon these studies by optimizing in vitro CRISPR specificity to isolate megabase-sized genomic segments by designing pairs of specific single guide RNAs (sgRNAs) to flanking DNA sequences. The intact DNA segment is released by in vitro CRISPR digestion and isolated via pulsed-field gel electrophoresis (PFGE). The isolated DNA segments are purified and directly sequenced using standard techniques, which can have >100-fold enrichment of the targeted megabase-sized regions. CISMR combines the specificity of in vitro CRISPR with the sensitivity of next generation sequencing, for a more direct, targeted, and affordable strategy to isolate and sequence complex and repetitive, megabase-sized regions of the genome to assess their biological significance.
MATERIALS AND METHODS
Preparation of agarose inserts
Agarose blocks of yeast genomic DNA and mouse genomic DNA are prepared similarly to previously described methods (8,9). Unless otherwise noted, cells are concentrated into 80 μl agarose blocks to yield approximately 8 μg of genomic DNA per block.
A fresh, single colony of Saccharomyces cerevisiae carrying the YAC clone ADK.D6 is grown in Ura-/Trp- synthetic complete liquid culture media until log-phase. Cells are rinsed twice by centrifugation at 1,000 g for 15 minutes at 4°C followed by resuspension of the pellet in 0.05 M EDTA (pH 7.5). Cells are counted on a hemocytometer and diluted to 6 × 109 cells/ml in 0.125 M EDTA (pH 7.5). An equal volume of cell suspension and 1.2% low melting point (LMP) agarose (Lonza) in 0.125 M EDTA (pH7.5) is mixed with 15.5 μl Zymolyase Solution (Supplementary Table S1) per ml of the mixture. 80 μl of the cell/agarose mixture is dispensed into each agarose block mold (Bio-Rad). Agarose blocks are stored at 4°C until the agarose mixture is solidified, ∼15 min. Solid agarose blocks are transferred into a conical tube and incubated with at least 2 volumes of 0.5 M EDTA and 7.5% beta-mercapthoethanol for 16 h at 37°C. This solution is replaced with a N-laurylsarcosine and Proteinase K (NDSK) buffer (Supplementary Table S1) and incubated another 16 h at 50°C.
For preparation of mouse genomic DNA in agarose blocks, a spleen is dissected from an adult female C57BL/6J mouse and immediately minced into 2 mm cubes in 2 ml 1× PBS in a sterile petri dish. The minced tissue and the 1× PBS is transferred to a sterile 15 ml glass dounce homogenizer and processed into single cells with ∼20 twisting strokes. Cells are transferred to a conical tube and rinsed twice by centrifugation at 1000 g for 15 min at 4°C followed by resuspension of the pellet in 5 ml 1× PBS. The cells are counted on the hemocytometer and diluted to 3 × 107 cells/ml in 1× PBS. This cell suspension is mixed 1:1 with 1.2% LMP agarose in 1× PBS and dispensed into 80 μl aliquots per agarose block mold. Agarose blocks are stored at 4°C until solidified, approximately 15 min. Solid agarose blocks are transferred to a conical tube and incubated with at least 2 volumes NDSK buffer for 40 h at 50°C.
Following NDSK incubation, agarose blocks of both yeast and mammalian genomic DNA are prepared identically for in vitro CRISPR. Agarose blocks are rinsed twice in 10–20 volumes of TE for 5 min at room temperature prior to a 16 h, room temperature incubation in 10–20 volumes of TE with 0.01 mM phenylmethylsulfonyl fluoride (PMSF) solution (Supplementary Table S1) with gentle shaking. Over the next 24 h, the blocks are rinsed several times in 10–20 volumes of TE with gentle shaking at 4°C. Agarose blocks are then stored long term at 4°C in TE or transferred into 1× Cas9 buffer (NEB, Supplementary Table S1) for a 2–16 h incubation with gentle shaking at 4°C prior to the CRISPR reaction.
Design and generation of sgRNAs
To improve specificity, 17 base sgRNA targets are used over the more traditional 20 base target sequences (10). Using both Target Finder (http://crispr.mit.edu) and ZiFIT Targeter software (11), single guide RNA (sgRNA) target sequences are designed from genomic regions of interest (Supplementary Table S2) using the mouse reference genome sequence (mm10) (12).
Two methods are used to produce in vitro transcription templates for sgRNAs: cloning target sequences into the pUC57-sgRNA plasmid vector (13) (Addgene) and PCR amplifying the pX458 plasmid vector (14) (Addgene) with forward primers containing both the T7 promoter sequence and the target sequences (Supplementary Table S2). For cloning into pUC57-sgRNA, it is first linearized with BsaI, then ligated with annealed target sequence oligonucleotides designed with overhanging tails: a 5′-TAGG-3′ tail on the PAM containing strand oligonucleotide and a 5′-AAAC-3′ tail on the reverse complement oligonucleotide. Clone inserts are confirmed by DNA sequencing. To terminate the in vitro transcription reactions on pUC57-sgRNA clones, the plasmids are linearized with DraI at a site immediately 3′ to the sgRNA sequence. In vitro transcription templates for all the Srsx sgRNAs are generated by PCR amplification, using the pX458 plasmid vector as a template, forward primers containing both the T7 promoter sequence and the target sequence, and a universal reverse primer designed to the 3′-end of the sgRNA sequence (Supplementary Table S2). Two-step PCR is performed with Phusion high-fidelity DNA polymerase following the manufacturer's recommendations (NEB). Reactions are cycled with a 98°C, 30 s hot start, followed by 35 cycles of 98°C for 30 s and 72°C for 20 s, and completed with one final 72°C extension for 1 min. Multiple reactions are performed and gel purified prior to in vitro transcription. All sgRNA templates are in vitro transcribed using the T7 polymerase based MEGAShortScript Kit (Ambion), and purified with the MEGAclear kit (Ambion) following the manufacturers’ recommendations. RNA products are sized on a standard 2% agarose gel, quantified by nanospectrometry, and diluted in nuclease-free water for long term storage at –80°C.
Digestion of genomic DNA via in vitro CRISPR
In vitro CRISPR digestions are performed on pure DNA templates and DNA imbedded in agarose blocks. Although sgRNAs and Cas9 are consistently used in a 1:1 molar ratio, total concentrations vary depending on the type of target DNA. SgRNAs and Cas9 are used in 10-fold molar excess of purified sources of target DNA, such as PCR products or plasmids. In a typical 30 μl reaction, 200 μM of each sgRNA is combined with 200 μM of Cas9 enzyme to digest 20 μM of purified target DNA. In this case, if two sgRNAs are used at 200 μM each, then the total sgRNA concentration of 400 μM is mixed with 400 μM of Cas9 enzyme. As determined empirically (Supplemental Figure S2), for optimal digestion of ∼2–10 μg genomic DNA in agarose blocks, sgRNAs and Cas9 are used at 40 nM in a 100 μl total reaction volume that contains the equivalent volume of 20–40 μl of an agarose block. This represents an >105 molar ratio of sgRNA/Cas9 to target genomic DNA. In this case, reaction components are typically concentrated in a 60 or 80 μl reaction mix with 1× Cas9 buffer and applied to a 40 μl (1/2) or 20 μl (1/4) slice of an agarose block, respectively, pre-equilibrated in 1× Cas9 buffer. In all types of in vitro CRISPR reactions, sgRNAs and the Cas9 enzyme are mixed with all the reaction components (1× Cas9 buffer and 1U/μl RNasin) except for the target DNA and pre-annealed in a 10 min incubation at 37°C. Once the target DNA is added, CRISPR reactions are incubated at 37°C for 1 h, unless otherwise specified. Cas9 only control reactions are carried through with all samples. Positive control reactions on YAC or BAC DNA are performed when possible. Reactions on agarose blocks are terminated by the replacement of the reaction mix with NDSK followed by at least a 1 h incubation with gentle shaking at 4°C. Reactions on purified DNA are terminated by the addition of 1/20 volumes of NDSK to the reaction followed by at least a 1 h incubation with gentle shaking at 4°C. PFGE of the reactions is typically performed immediately, but terminated reactions can be stored overnight at 4°C.
Pulse field gel electrophoresis (PFGE) conditions
For optimal resolution of DNA by PFGE, each lane is loaded with an intact slice (20–40 μl) of an agarose block, containing approximately 2–10 μg of genomic DNA (15). Agarose blocks are mounted directly on the bottom edge of the gel comb and incorporated into a 14 × 21 cm gel poured with 150 ml tempered, high-strength agarose (Aqua-Por). Megabase resolution gels are 1% agarose in 0.5× TBE (Bio-Rad). Multi-megabase resolution gels are 0.8% agarose in 1× TAE (Bio-Rad). Size ladders of yeast chromosomes or the Yeast Chromosome PFG Markers (Bio-Rad) are loaded on all megabase resolution gels. H. wingei CHEF DNA Size Markers (Bio-Rad) are loaded on all multi-megabase resolution gels. Using a glass coverslip, all raised edges of the agarose gel are trimmed away to allow optimal buffer recirculation across the gel during electrophoresis. All PFGE is performed on a Bio-Rad CHEF-DR III system with an external chiller and recirculation pump. The PFGE apparatus and ∼2 L of running buffer is chilled to 14°C prior to and during the run. Megabase resolution PFGE is carried out at a 120° angle and 5 V/cm for 66 h with switch times ramping between 47 and 170 s (15). Multi-megabase resolution PFGE is carried out at a 106° angle and 3 V/cm for 48 h with a fixed switch time of 500 s. Gels are post-stained for 1 h in 1× Diamond Stain (Promega) at room temperature in running buffer adjusted to pH 7.7. Images are captured and evaluated on a Bio-Rad Gel Doc™ XR+ with Image Lab™ Software equipped with an XcitaBlue™ conversion screen and filter kit (Bio-Rad).
Southern blotting
When necessary, DNA resolved on PFGE gels is transferred to a membrane and hybridized with a probe specific for the targeted DNA segment. Briefly, gels are bathed, with gentle rotation, in 0.25 M HCl for 30 min followed by 0.4 M NaOH for 20 min. Standard Southern blotting with vertical transfer of the DNA is performed overnight in 0.4 M NaOH onto a charged nylon membrane, Amersham Hybond-N+ (GE). The membrane is air-dried and pre-hybridized and hybridized in High SDS Buffer (Roche, Supplementary Table S1) sealed in a rotating glass bottle incubated in a hybridization oven. A PCR-DIG-labeled probe (Supplementary Table S2) is prepared with the PCR DIG Probe Synthesis Kit (Roche) according to the manufacturer's recommendations, denatured and added to the hybridization for an overnight incubation at the calculated temperature of hybridization (TH) (Roche, Supplementary Table S2). The membrane is washed two times at low stringency, and two times at high stringency followed by detection of the DIG-labeled probe by the chemiluminescent assay with Disodium 3-(4-methoxyspiro {1,2-dioxetane-3, 2′-(5′-chloro)tricyclo [3.3.1.13,7]decan}-4-yl)phenyl phosphate (CSPD) alkaline phosphatase substrate following the manufacturer's recommendations (Roche). X-ray film is exposed to the blot overnight.
Next generation sequencing library preparation and sequencing
In order to sequence DNA segments resolved by PFGE, DNA segments are excised from the gel, sonicated and purified for Illumina-sequencing library construction. Up to 10 high DNA concentration (5–10 μg) slices of agarose blocks are digested in individual 100 μl in vitro CRISPR reactions (Otc, Ssx or Srsx, Supplementary Table S2) as described above. The collection of agarose blocks for each in vitro CRISPR reaction are aligned on a preparative comb for the PFG and resolved by electrophoresis as described above. Sterile glass coverslips are used to excise DNA segments from the gel. Based on the weight, each gel slice is dissolved at 55°C in the appropriate volume of Binding Buffer as described and provided by the GeneJET Gel Extraction Kit (Thermo Scientific). To shear the DNA segments into 200–500 bp fragments, each liquified gel slice is subjected to 12 cycles of sonication on the Fisher Scientific™ Model 505 Sonic Dismembrator equipped with a 0.32 cm probe at 4°C set to 25% amplitude with 20 s of sonication followed by 40 s rest. The sonicated gel solution is then bound to a GeneJet column and purified as described by the manufacturer (ThermoFisher). DNA from each column is eluted in 30 μl of 10 mM Tris (pH 8.5). If multiple columns are needed for gel purification, then elutions are combined and dried down into a 60 μl total volume. The concentration of the purified DNA fragments is estimated by applying 10 μl of the sample to High Sensitivity DNA Qubit quantitation (Qubit).
Since the quantity of gel purified DNA fragments is limited, we use the NEBNext UltraII DNA Library Prep kit, optimized for low-DNA-input, to generate Illumina-sequencing libraries. The total yield from the sonication and gel purification of each DNA segment, ranging from 3.1 to 7.2 ng (Supplemental Table S3), is used in the library protocol as described by NEB. Size selection following adapter ligation is bypassed. The minimal number of PCR cycles, recommended by the NEBNext Ultra II protocol, is used in an attempt to achieve a 100 ng library yield. Barcoded paired-end 75nt Illumina (version 3) sequencing was performed on a MiSeq following the manufacturers recommendation (Illumina).
Sequence analyses
Paired-end Illumina reads are processed for quality control using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc) and single end reads (forward) are mapped to the Mus musculus genome (mm10) using Bowtie2 (16) requiring perfect match sequence alignments (–score-min C, 0, -1). The resulting BAM files are used to perform fold enrichment and on-target bases estimates using PICARDs HsMetrics (https://broadinstitute.github.io/picard/), where the number of sequencing reads present in the target sequence is compared to non-targeted genomic sequence. Target regions for the 263 kb, 610 kb and ∼2.3 Mb mouse X chromosomal regions are defined by the sequence intervening the two sgRNA PAM sites (Supplementary Table S2). Bedgraphs are generated using Bedtools (17) and visualized in IGV Viewer (18).
RESULTS
To optimize and verify the specificity of CISMR, we targeted a 263 kb segment of the mouse X chromosome carried on a yeast artificial chromosome (YAC) clone, ADK.D6 (19), carried in Saccharomyces cerevisiae. Native yeast chromosomes and intact CRISPR-targeted DNA digestion products are readily resolved as single segments upon PFGE (15). We designed sgRNAs to truncated, 17-nt, target sequences (10) flanking a 263 kb segment of the mouse X chromosome containing the Otc locus (Supplementary Figure S1, Supplementary Table S2). Agarose blocks with intact yeast genomic DNA were exposed to varying concentrations of sgRNA/Cas9 in overnight in vitro CRISPR reactions. Digested yeast genomes show clear, intact, yeast chromosomes and the predicted sizes of DNA segments of the ADK.D6 YAC clone (Figure 1A, Supplementary Figure S1). The in vitro CRISPR reaction is highly specific, since over 12 Mb of endogenous yeast genomic DNA remains primarily uncut. The extent of in vitro CRISPR digestion of the YAC is dependent on the sgRNA/Cas9-concentration (Supplementary Figure S2). Complete cutting of the targeted YAC clone is achieved with 40 nM of sgRNA/Cas9 (Supplementary Figure S2). This represents a >105 molar ratio of sgRNA/Cas9 to target yeast genomic DNA. Increases >40 nM do not improve digestion efficiency, since this is most likely the concentration required to saturate the volume of the agarose block. Since CRISPR sequence-specificity increases with lower incubation times (20), we next optimized the specificity of in vitro CRISPR reactions by minimizing CRISPR digestion time. A time course ranging from 1 to 16 h shows maximal digestion is achieved after a 1-h incubation (Supplementary Figure S3).
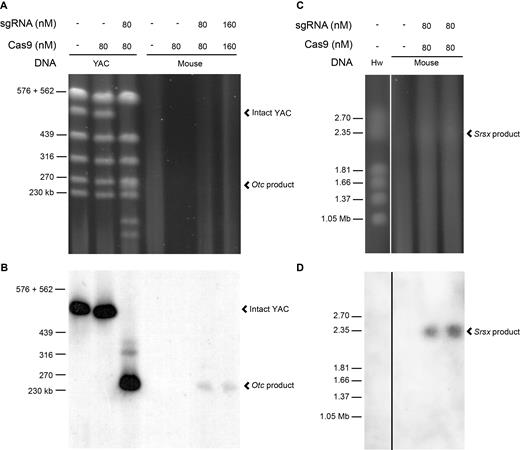
Large segments of the mouse X chromosome are specifically targeted with in vitro CRISPR. (A) The specific products of the in vitro Otc CRISPR digestion of both YAC ADK.A6 DNA and mouse genomic DNA are resolved by PFGE and detected with Diamond stain (Promega) (Supplementary Figure S1). Whereas the expected YAC digestion products are clearly detectable, digestion of the more complex mouse genomic DNA does not produce detectable products. Untreated and Cas9-only digestions of YAC and mouse genomic DNA serve as negative controls. (B) A Southern blot of the PFG shown in (A) is hybridized with a DIG-labeled Otc probe. The intact YAC chromosome (∼520 kb) and the expected 263 kb in vitro Otc CRISPR digestion product from both YAC and mouse genomic DNA are visible upon chemiluminescent detection of the probe and exposure of X-ray film. (C) A faint but, detectable ∼2.3 Mb in vitro Srsx CRISPR digestion product from mouse genomic DNA is resolved by PFGE along with the multi-megabase H. wingei (Hw) chromosomes ladder (Bio-Rad) labeled on the left. (D) The Southern blot of the PFG shown in (C) is hybridized with a DIG-labeled Srsx probe. The estimated ∼2.3 Mb in vitro Srsx CRISPR digestion product from mouse genomic DNA is visible upon chemiluminescent detection of the probe and exposure of X-ray film.
We applied YAC-optimized CISMR to mouse genomic DNA, using the same pair of sgRNAs flanking the 263 kb mouse X chromosome region. As expected, upon PFGE and post-staining, a 263 kb segment is not detectable in PFGE lanes of digested mouse genomic DNA due to the limited number of mammalian genome copies in approximately 4 μg of genomic DNA (Figure 1A). In addition, inherent to the large size of mammalian chromosomes, preparations of intact mammalian genomic DNA in agarose blocks results in some DNA shearing and is seen as a faint smear in all lanes, including the undigested control lanes of the PFGE (Figure 1A). However, upon Southern blotting of the PFG and hybridization with a probe specific to the targeted 263 kb DNA segment, we detect a single, specific segment at the expected 263 kb size (Figure 1B).
To expand the size range of isolated DNA segments using CISMR, we used the mouse X chromosome reference sequence to design pairs of sgRNAs to sequences flanking both the 610 kb region containing the Ssx gene family and the 3.4 Mb region containing the Srsx gene family (Supplementary Figure S1, Supplementary Table S2). Because the reference sequence of the Srsx region remains incompletely assembled, the size of the region is only an estimate. Surprisingly, large DNA segments for both Ssx and Srsx could be visualized directly upon staining the pulsed field gels (Figure 1C, Supplementary Figure S4A, Supplementary Figure S5). To ensure these DNA segments contain the targeted regions, we hybridized Southern blots of the PFGs with probes specific to the Ssx or Srsx sequence, producing positive signals for the 610 kb segment (Supplementary Figure S4B) and ∼2.3 Mb segment (Figure 1D), respectively. The ∼2.3 Mb in vitro Srsx CRISPR segment, 1 Mb smaller than predicted by the mouse X chromosome reference sequence, is consistent with a previous estimate of its size (21) and in triplicate experiments appears consistently via PFGE (Supplementary Figure S5). CISMR is capable of generating specific, intact genomic DNA segments of various sizes, in the presence of over 3 billion bases of non-specific (‘off-target’) mammalian genomic DNA sequence.
To determine the extent of enrichment by CISMR, the three mouse X chromosome DNA segments were purified from preparative PFGs and sequenced via Illumina sequencing. Visualization of read coverage to the three regions shows a heavy enrichment of target sequence as compared to flanking sequence (Figure 2A, Supplementary Figure S6). Additionally, the enrichment of sequencing reads ends exactly at the sgRNA targeted digestion site (Figure 2B, Supplementary Figure S6C). A quantitative enrichment analysis of reads mapping to the targeted sequence as compared to the rest of the genome, shows the 263 kb, 610 kb and ∼2.3 Mb segments are enriched 75-fold, 174-fold and 39-fold, and have on-target bases percentages of 0.7, 3.9 and 4.9, respectively (Supplementary Table S3). Triplicate experiments isolating the ∼2.3 Mb segment yielded 3.1, 4.1 and 7.0 ng and sequencing of the triplicate samples shows the fold enrichment (39, 77 and 86) and on-target bases percentages (4.9, 9.5 and 10.7) are reproducible (Supplementary Table S3). Differences in enrichment and on-target bases between the different size DNA segments could be due to the incomplete assembly of the reference sequence for the genomic region (e.g. Srsx region) or inherent variation of sheared DNA co-purifying with DNA segments on PFGs. Sequencing of the large in vitro CRISPR isolated DNA segments shows that CISMR is precise, specific and efficient.
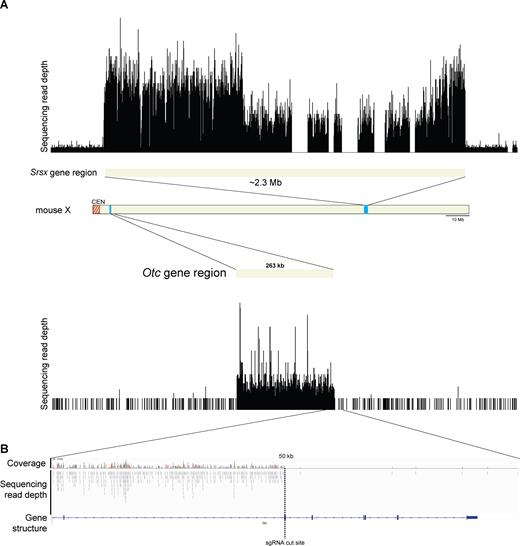
Enrichment of Illumina sequencing reads of the isolated 263 kb and ∼2.3 Mb genomic segments. (A) Single-end sequencing read depth of the ∼2.3 Mb Srsx-gene array region and the 263 kb Mb Otc-gene region, both of which are flanked by 500 kb of non-targeted sequence. The five gaps present within the Srsx region are due to incomplete assembly of the reference sequence. Schematic of the mouse X chromosome shows the origin of the targeted and sequenced genomic DNA segments. (B) Higher resolution of Illumina sequencing read depth and coverage across 25 kb of sequence flanking the 3′ Otc-gene region sgRNA cut site (vertical dotted line). The sgRNA targets exon 5 of Otc, which is consistent with the alignment of multiple reads up to the sgRNA cut site.
DISCUSSION
CISMR is a novel, cost-effective method to decipher specific complex, megabase-sized genomic regions across a diversity of individuals, for any region of the genome. We envision the incorporation of CISMR into long-read sequencing approaches and the generation of region-specific clone libraries, both of which are compatible with low inputs of DNA. Combining CISMR with long-read sequencing approaches and region-specific clone library generation will both improve the accuracy of reference sequence assemblies by resolving sequence gaps in any genome (22) and enable a more complete assessment of genomic variation within structurally complex regions.
We can envision combining CISMR with long-read sequencing technologies and SHIMS (Single-Haplotype Iterative Mapping and Sequencing) to more efficiently and cost-effectively resolve complex genomic regions comprised of segmental duplications (Figure 3). The choice of sequencing strategy is dependent on whether the segmental duplications are large and highly identical (>10kb and >96% nucleotide identity) (23) or short with low levels of sequence identity (low identity). To accurately resolve low identity segmental duplications, CISMR can be combined directly with long-read sequencing technologies, including Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT). Whole-genome PacBio sequencing was used to generate a high-quality assembly of the gorilla genome (24) and a human haploid genome (25,26), resolving low identity segmental duplications previously intractable with short-read sequencing technologies. However, to study structural variation of segmental duplications of a specific megabase-sized region via whole genome PacBio sequencing is costly and time consuming. Combining CISMR with long-read sequencing technologies will enable the rapid and accurate resolution of many complex low identity segmentally duplicated regions throughout the genome at a fraction of the cost.
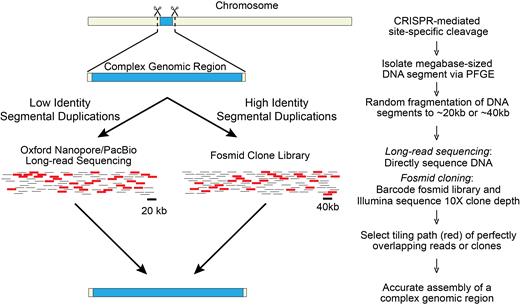
Combining CISMR with sequencing strategies to resolve megabase-sized structurally complex regions of the genome. CISMR isolated DNA segments containing low identity or high identity segmental duplications can be resolved with current long-read sequencing technologies or region-specific clone libraries, respectively. Both approaches are amenable to low inputs of DNA to generate an accurate assembly of the complex genomic region.
To accurately resolve high identity segmental duplications, long-read sequencing approaches fall short because of high error rates, thus requiring CISMR to be combined with the clone-based SHIMS approach. SHIMS has been used to generate high quality assemblies of nearly-identical segmental duplications (amplicons) of haploid Y chromosomes, across multiple species over many years (4,27–31). However, SHIMS is traditionally dependent on whole genome clone libraries making it an expensive, labor-intensive, and time-consuming approach. Instead of using whole-genome clone libraries to accurately sequence and resolve megabase-sized ampliconic regions (3,4), we envision CISMR will be used to prepare region-specific fosmid or BAC libraries from continuous, targeted megabase-sized segments (Figure 3). Fosmid or BAC-based cloning is preferable to yeast-based transformation assisted recombination (TAR) cloning (32), because amplicons can undergo rearrangements when inserted into yeast artificial chromosomes (33). 10× coverage of fosmid clones will be sequenced via Illumina to fully resolve their underlying sequence and ordered and oriented across the region. CISMR, when combined with SHIMS, will provide a more efficient and cost-effective approach to sequence megabase-sized complex regions of the genome with highly identical segmental duplications.
CISMR provides a new method to rapidly assess variation across a diversity of megabase-sized complex genomic regions such as telomeres, centromeres, rDNA arrays and gene amplifications (e.g. HER2 expansions in Breast cancer) (34,35). CISMR is distinct from other targeted sequencing methods (e.g. hybrid capture), because it preserves the integrity and continuity of megabase-sized DNA segments and does not require knowledge of the underlying sequence. CISMR can also be used for genetic mapping purposes, similar to RecA-assisted restriction endonuclease (RARE) cleavage (36), but unlike RARE-cleavage CISMR does not require knowledge of the intervening sequence and is not dependent on the location of restriction sites. Further development of CISMR may make the method even more efficient. For example, CISMR is amenable to multiplexing excision reactions for the isolation of DNA segments at several sizes. CISMR isolated DNA segments can also be sequenced using any of the next generation sequencing technologies, allowing customized approaches to study the underlying sequence. Overall, the universality of CISMR will provide opportunities to generate novel insights into the biological roles of complex, megabase-sized regions of the genome across species.
DATA AVAILABILTY
All Illumina next generation sequencing libraries can be found in the NCBI short read archive (SRA) under accession numbers: SRR5380205–7 and for the replicates under SRR5639072–3.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR online.
ACKNOWLEDGEMENTS
We thank Juan Lucas Argueso, David Burke, Sarah Emery, and Jeffrey Kidd for technical advice and John Moran for use of his MiSeq. We also thank Winston Bellott, David Burke, Jeffrey Kidd and John Moran for discussions and comments on the manuscript.
FUNDING
Research reported in this publication was supported by NICHD of the National Institutes of Health [HD064753]. Funding for open access charge: University of Michigan.
Conflict of interest statement. None declared.
REFERENCES

{kind=link}
{kind=link}
{kind=link}
Comments