
Abstract
Objective. To evaluate the safety and efficacy of ocrelizumab (OCR) in Japanese patients with rheumatoid arthritis (RA) with an inadequate response to methotrexate (MTX).
Methods. RA patients with an inadequate response to MTX 6–8 mg/week received an infusion of 50, 200, or 500 mg OCR or placebo on Days 1 and 15 and were observed for 24 weeks. The double-blind period was prematurely terminated because of a possible risk for serious infection from OCR.
Results. A total of 152 patients were randomized into the study. The incidence of infection was 37.7% (43/114) in the OCR groups combined, compared to 18.9% (7/37) in the placebo group. Serious infections occurred in 7 patients in the OCR groups combined; there were no serious infections in the placebo group. Among the serious infections, Pneumocystis jirovecii pneumonia occurred in 2 patients in the OCR 200 mg group. The American College of Rheumatology 20% response rates at Week 24 (the primary endpoint) of the OCR 50, 200, and 500 mg groups were 54.1% (p = 0.0080), 55.6% (p = 0.0056), and 47.2% (p = 0.044), respectively, all significantly higher than that of the placebo group (25.0%).
Conclusion. These results suggest inappropriate benefit-risk balance of OCR in this patient population. Because rituximab is not approved for treatment of RA in Japan, it will be necessary to investigate safety and efficacy of other anti-B cell therapies in Japanese patients with RA. (ClinicalTrials.gov NCT00779220).
- OCRELIZUMAB
- RHEUMATOID ARTHRITIS
- B CELL DEPLETION
- CLINICAL TRIALS
The possible involvement of B cells in the pathogenesis and progression of RA, including autoantibody production, autoantigen presentation, T cell activation, and production of proinflammatory cytokines and chemokines, has been suggested1,2,3,4,5,6. Based on these reports, clinical trials of rituximab (RTX), a chimeric anti-CD20 monoclonal antibody (mAb) targeting CD20 molecules, were conducted in patients with rheumatoid arthritis (RA)7,8. Subsequently, RTX was approved for treatment of RA in Europe and the United States.
Ocrelizumab (OCR) is a humanized mAb that also targets CD209,10 and eliminates B cells by inducing antibody-dependent cell-mediated cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC), and apoptosis. While the epitopes recognized by OCR and RTX on the extracellular domain of the CD20 molecule partially overlap, OCR offers some advantages over RTX. First, OCR is expected to be better tolerated over repeated and longterm administration because OCR induced higher ADCC activity and lower CDC activity than RTX in vitro; this has clinical relevance because CDC activation has been associated with the incidence and severity of infusion-related reactions (IRR)11. Second, as a humanized mAb, OCR may have lower immunogenicity than RTX, a chimeric mAb.
A 6-month, double-blind, phase I/II study of OCR (the ACTION study) was undertaken in the United States, enrolling patients with RA with an inadequate response to disease-modifying antirheumatic drugs (DMARD). The results of the ACTION study confirmed the clinical usefulness of OCR in combination with methotrexate (MTX)12. To investigate the dose-responsive effects of OCR in Japanese patients with RA, we conducted a 24-week, placebo-controlled, double-blind, phase II study of OCR with concomitant MTX treatment in Japanese patients with RA whose response to MTX had proved inadequate.
MATERIALS AND METHODS
Patients
Our study was conducted at 37 sites in Japan with approval from the Institutional Review Board at each participating site. Written informed consent was obtained from each patient participating in the trial. Our study was conducted in accord with the Declaration of Helsinki and the Good Clinical Practice guidelines, and was registered at ClinicalTrials.gov, NCT00779220.
Patients selected were ≥ 20 years old, fulfilled the American College of Rheumatology (ACR) 1987 revised criteria for RA13, were rheumatoid factor (RF)-positive (> 20 IU/ml), showed an inadequate response to MTX at a dosage of 6–8 mg/week (maximum approved dose in Japan at that time: 8 mg/wk) for at least 12 weeks with a stable dose for the last 4 weeks before study treatment, had not used tocilizumab, infliximab, adalimumab, or leflunomide for at least 8 weeks before study treatment, and had used no other DMARD except MTX for at least 4 weeks before study treatment. Active disease was defined as swollen joint count ≥ 8 (66-joint count), tender joint count ≥ 8 (68-joint count), and either serum C-reactive protein (CRP) ≥ 1.5 mg/dl or erythrocyte sedimentation rate (ESR) ≥ 28 mm/h. Key exclusion criteria were additional autoimmune disorders, previous treatment with cell-depleting agents, neutrophil count < 1500/μl, platelet count < 100,000/μl, IgG or IgM less than the lower limit of normal (LLN), or hemoglobin < 8.5 g/dl.
Study design
This was a placebo-controlled, double-blind, multicenter, phase II study. The overall study design is illustrated in Figure 1. The subjects were randomly allocated into 4 groups, the OCR 50, 200, or 500 mg group, or the placebo group, in equal numbers and then given an infusion of their assigned investigational product on Days 1 and 15. Methylprednisolone 100 mg was given intravenously as premedication 30 min before administration of each investigational product. The use of oral antihistamine and acetaminophen 30 to 60 min before administration of investigational product was also permitted. Patients who were withdrawn from the double-blind period entered the safety followup period and were followed for at least 48 weeks from the first infusion of investigational product. This report includes the initial 24-week results.

The study design. †Use of additional treatments for RA was permitted after Week 8 if control of disease activity was inadequate, at the discretion of the investigators or subinvestigators. OCR: ocrelizumab; MTX: methotrexate.
All patients received uninterrupted stable dosages of MTX (6–8 mg/wk) and folate (≥ 5 mg/wk) from at least 4 weeks before the initiation of study treatment to the end of the study period. Concomitant use of a stable dosage of oral corticosteroid (prednisolone equivalent dose ≤ 10 mg/day) was permitted if the dosage was unchanged in the last 4 weeks before the study, and the concomitant use of nonsteroidal antiinflammatory drugs was also permitted if the dosage had not been changed within the last 2 weeks. Concomitant use of biological or nonbiological DMARD other than MTX was prohibited. The following rescue treatments were allowed from Week 8 at the investigator’s discretion if control of disease activity was judged inadequate: increased MTX up to 8 mg/week, use of nonbiological DMARD, increase of oral corticosteroid, intraarticular administration of corticosteroid, intraarticular administration of hyaluronic acid preparation, and the use of 1 biological DMARD (excluding RTX).
Evaluation
Safety and efficacy were evaluated on Days 1 and 15, and every 4 weeks thereafter from Week 4 to Week 24 in the double-blind treatment period. During the safety followup period, safety and efficacy were evaluated every 12 weeks. The primary efficacy endpoint was the ACR 20% (ACR20) response rate at Week 2414. The ACR50 and ACR70 response rates and a reduction in the Disease Activity Score (DAS28-ESR) values15 and European League Against Rheumatism (EULAR) response rates16 over time up to Week 24 were calculated as secondary endpoints. The percentage of patients achieving DAS28-ESR remission (DAS28-ESR < 2.6) by Week 24 was investigated as exploratory analyses.
To evaluate safety, all adverse events (AE) that occurred during the study were recorded; their severity was judged using the National Cancer Institute (NCI) Common Toxicity Criteria (CTC) Version 3.0. Serious AE (SAE) were defined using criteria from the International Conference on Harmonization. Serious infections (SI), defined as SAE infections or infections requiring intravenous antibiotic injection, were tabulated. Human anti-human antibody (HAHA) and serum immunoglobulin (IgG, IgM, and IgA) concentrations were also measured. To evaluate pharmacokinetics, OCR concentration in serum was measured, and the number of CD19-positive cells in peripheral blood was measured using flow cytometry.
Statistical analyses
The target sample size was calculated based on the ACR20 response rate in the ACTION study. Using an allocation ratio of 1:2 (placebo vs combined OCR 200 and 500 mg groups), the ACR20 response rate in the combined OCR group of 48.7% and the placebo group of 24.3%, a 2-tailed significance level of 5%, and a power of 80%, the required sample size was calculated by the chi-square test to be 46 patients per group. Allowing for untreated patients, the target group size was set at 50 patients, giving a total target sample size of 200 patients. Calculation of the sample size was performed using nQuery Advisor Version 5.0 (Statistical Solutions Ltd., Farmer’s Cross, Ireland).
While our study was in progress, an increased incidence of SI, including opportunistic infections, was reported in multinational clinical trials of OCR that were being conducted at the same time. Based on these safety reports, the enrollment of new patients and the administration of the investigational product in our study were halted, resulting in administration of investigational product to only 151 patients. The double-blind period was prematurely terminated in January 2010 and all patients entered a safety followup period.
The analysis of efficacy was performed using 145 patients (36 patients in the placebo group, 37 in the OCR 50 mg, 36 in the OCR 200 mg, and 36 in the OCR 500 mg), excluding 1 patient in the placebo group, 2 in the OCR 50 mg, and 3 in the OCR 200 mg group who did not receive the second infusion of investigational product because the study was stopped. We recalculated the statistical power and confirmed that it decreased from 80% to 69% with the same assumptions except for the number of patients. The analysis of safety was performed using 151 patients who received investigational products at least once. Safety data were evaluated up to 24 weeks from the first infusion of investigational product regardless of whether patients completed the double-blind period.
The ACR20 response rate at Week 24 (primary endpoint) and ACR50 and ACR70 response rates at Week 24 (secondary endpoints) in each OCR group were compared with the placebo group using the Cochran-Mantel-Haenszel test, accepting a 2-sided significance level of 5%. Based on the predefined analysis plan, descriptive statistics were calculated for the remaining endpoints, but no intergroup comparisons were performed. Adjusted mean changes in DAS28-ESR were based on the analysis of covariance using the baseline value as a covariate.
Efficacy data obtained after the day of rescue treatment or after the day when the decision to withdraw was made were handled as follows: categorical data (ACR responses, EULAR response rates, DAS28-ESR remission) were treated as “no response,” continuous data (DAS28-ESR) as “missing data,” and the last observation was carried forward.
RESULTS
Baseline characteristics and patient distribution
The mean RA disease duration of the patients in each group was 6.7–10.0 years. The patients had high RA disease activity with a mean DAS28-ESR of 6.3–6.5, a mean serum CRP level of 1.8–3.0 mg/dl, a mean ESR of 53.1–57.0 mm/h, and functional disabilities shown by a mean J-HAQ of 1.3–1.4. The mean MTX dosage was 7.3–7.6 mg/week. In each group, 25.6%–38.9% of the patients had previously received a biological DMARD (Table 1).
Rheumatoid arthritis (RA) patient demographics and baseline disease characteristics (n = 151).
Including withdrawals because of the halt in administration of the investigational product, the patients who withdrew from the study before Week 24 numbered 6 in the placebo group, 10 in the OCR 50 mg, 11 in the OCR 200 mg, and 6 in the OCR 500 mg groups. The number of patients who withdrew because of insufficient response was 3 in the placebo group and none in the OCR groups. The proportion of patients receiving rescue treatments up to Week 24 was 32.4% in the placebo group, but lower in the OCR groups: 12.8% in the OCR 50 mg, 7.7% in the OCR 200 mg, and 16.7% in the OCR 500 mg groups (Figure 2).

Disposition of patients with RA at Week 24. *Dosing of investigational product was stopped, patients were withdrawn from the study, and enrollment of new patients was halted because of the increased incidence of serious infections, including opportunistic infections, reported in other multinational clinical studies of ocrelizumab (OCR). AE: adverse event.
Safety
During the 24-week observation period, the incidence of AE was 59.5% in the placebo group and 79.5% in the OCR 50 mg, 79.5% in the OCR 200 mg, and 61.1% in the OCR 500 mg groups (Table 2). The majority of AE were infections and IRR. An IRR was defined as an AE occurring during or within 24 hours after administration of investigational product.
Summary of adverse events (AE) in the safety analysis population of patients with rheumatoid arthritis (n = 151) during the 24-week observation period. Values are the number (%) of patients.
The proportion of subjects experiencing at least 1 infection was 18.9% (7/37) in the placebo group and 37.7% (43/114) in the OCR groups combined. There were no patients with SI in the placebo group and 7 in the OCR groups combined. There were 4 SI in 2 patients in the OCR 50 mg group, consisting of 1 incident each of herpes zoster, pneumonia, sepsis, and septic shock. Six SI occurred in 4 patients in the OCR 200 mg group, 2 incidents of Pneumocystis jirovecii pneumonia (PCP), and 1 each of sepsis, herpes simplex, bacterial pneumonia, and febrile neutropenia. There was 1 SI (epididymitis) in 1 patient in the OCR 500 mg group.
Two incidents of malignant tumors (uterine cancer and ovarian cancer) in 1 patient in the OCR 500 mg group were reported, which were diagnosed 153 days after the first infusion of OCR. There were no intergroup differences in the incidences of other AE.
There were 2 deaths during our study. One was a 61-year-old man who had concurrent depression and hypertension, and a history of cerebral infarction and cerebral hemorrhage. He developed pneumonia and sepsis 67 days after administration of OCR 500 mg followed by septic shock, disseminated intravascular coagulation, and multiorgan failure and died the following day. The other death was a 64-year-old man in the placebo group; he died of acute respiratory failure after withdrawal from the study because of insufficient response. No definitive diagnosis was made and an autopsy was not performed.
The increase in incidences of IRR following the first administration (Day 1) of investigational product was dose-dependent: 0% in the placebo group and 15.4% in the OCR 50 mg, 20.5% in the OCR 200 mg, and 25.0% in the OCR 500 mg groups. Following the second administration (Day 15) of investigational product, the incidence of IRR was markedly decreased in all 3 OCR groups (Table 2, Figure 3), 2.9% in the OCR 50 mg, 6.1% in the OCR 200 mg and 8.8% in the OCR 500 mg groups. All patients, except for 1 in the OCR 500 mg group, who experienced an IRR at the second administration also had an IRR at the first administration. Of the 26 IRR, 4 were moderate (NCI CTC Grade 2) and 22 were mild (NCI CTC Grade 1). One patient in each of the OCR 200 mg and the OCR 500 mg groups withdrew from the study because of IRR.

Incidence of infusion-related reactions at Days 1 and 15. Patient numbers shown here represent patients in each group at Days 1 and 15. OCR: ocrelizumab.
No placebo group patients became HAHA-positive during the study, but 2 patients in the OCR 50 mg group and 1 each in the OCR 200 mg and OCR 500 mg groups were HAHA-positive. An IRR occurred in 1 of the 4 patients who became HAHA-positive; this patient developed the IRR at the first administration prior to the expression of HAHA. No serious AE occurred in any HAHA-positive patient. Two of the 4 HAHA-positive patients achieved ACR20 at Week 24, and 1 achieved ACR70. A comparison of HAHA-positive and HAHA-negative patients showed no consistent difference in serum OCR concentration during the study period. The presence of HAHA did not appear to influence either efficacy or safety outcomes.
Efficacy
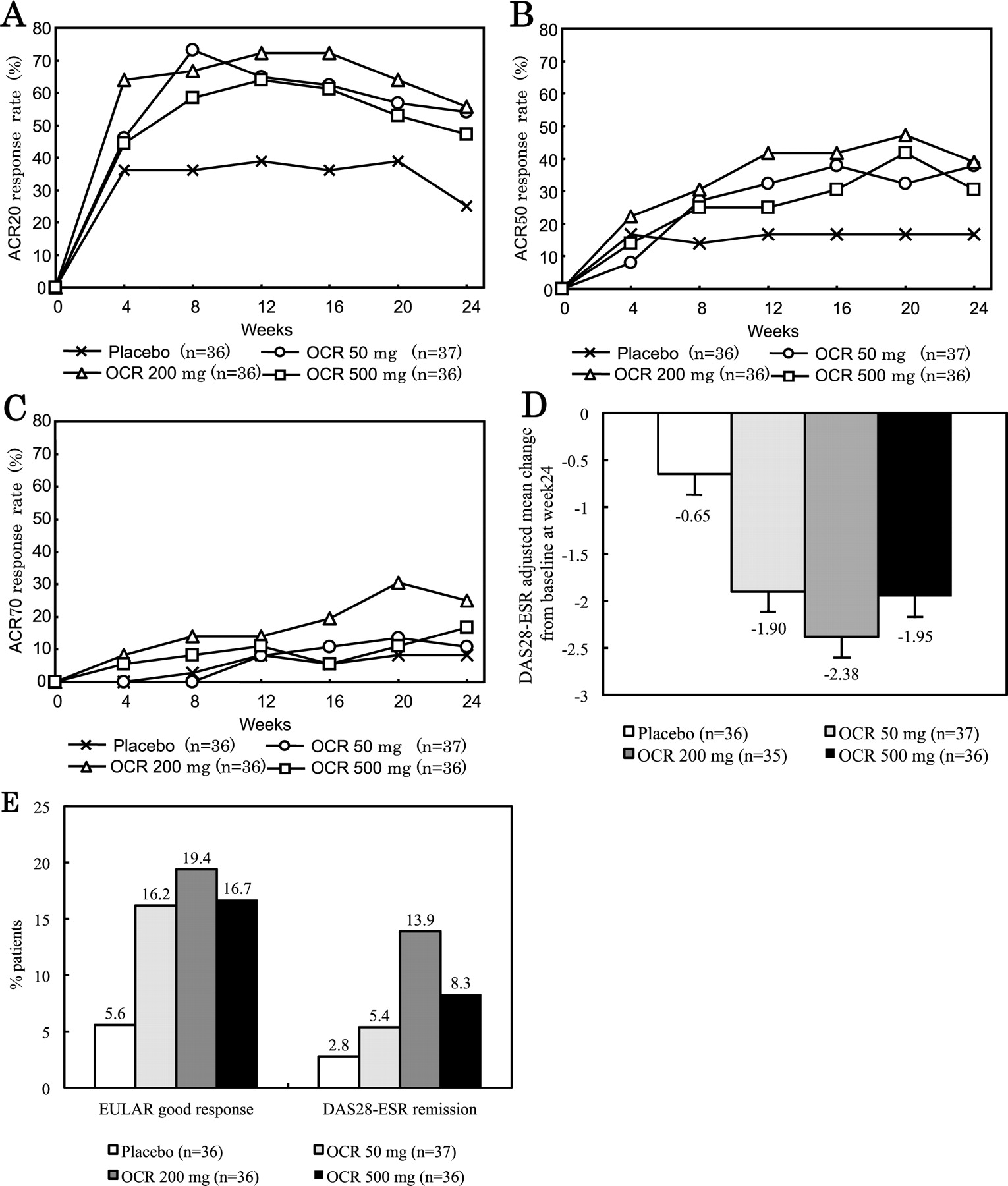
The ACR20 response rates at Week 24, the primary endpoint, in the OCR groups were significantly higher than the 25.0% of the placebo group [OCR 50 mg: 54.1% (p = 0.0080), OCR 200 mg: 55.6% (p = 0.0056), OCR 500 mg: 47.2% (p = 0.044)]. The ACR50 responses at Week 24 were 16.7% for the placebo group, 37.8% for the OCR 50 mg, 38.9% for the OCR 200 mg, and 30.6% for the OCR 500 mg groups. The ACR50 response rates in the OCR 50 mg and OCR 200 mg groups were significantly higher than those in the placebo group (p = 0.038, p = 0.031, respectively). The ACR20, ACR50, and ACR70 response rates over time are shown in Figure 4 A–C. The adjusted means (± SE) of the ΔDAS28-ESR, the good responses rates using the EULAR response criteria, and the DAS28-ESR clinical remission rates (DAS28-ESR < 2.6) of the OCR groups at Week 24 were better than those of the placebo groups (Figure 4D, 4E).

Clinical efficacy of ocrelizumab (OCR). A. ACR20 response rate over time. B. ACR50 response rate over time. C. ACR70 response rate over time. Patients receiving rescue therapy or withdrawing from the study were classified as nonresponders. D. DAS28-ESR mean changes from baseline at Week 24. Error bars represent standard error of the mean. E. The proportion of patients achieving a good response according to the EULAR criteria and remission according to DAS28-ESR. ACR: American College of Rheumatology; DAS28: Disease Activity Score (28 joint count); ESR: erythrocyte sedimentation rate; EULAR: European League Against Rheumatism.
Pharmacodynamics
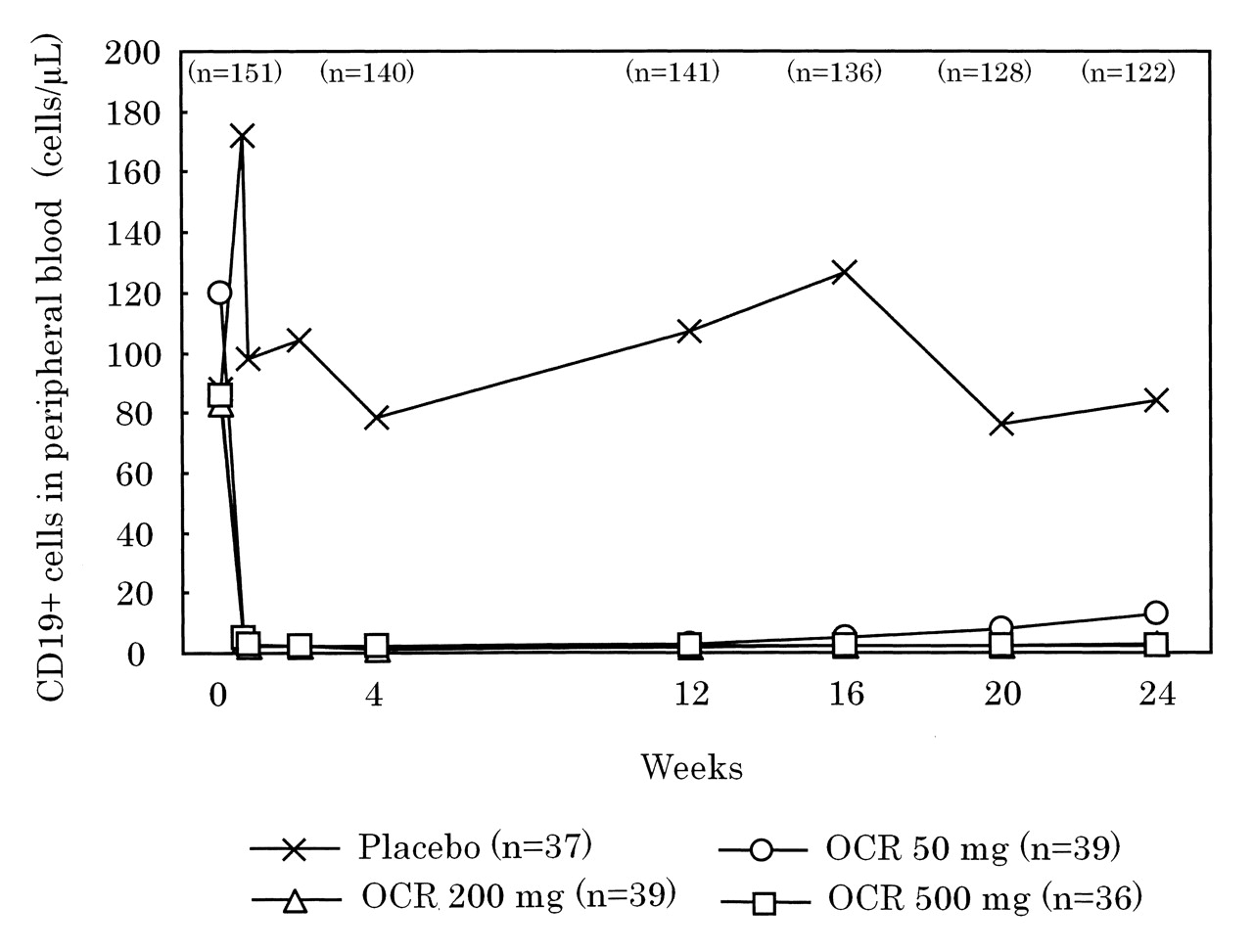
Although the number of CD19-positive cells increased transiently in the placebo group following intravenous administration of methylprednisolone as premedication on Days 1 and 15, the number remained stable through Week 24. In all 3 OCR-treated groups, the number of CD19-positive cells in peripheral blood decreased rapidly after the first administration of OCR; that effect was maintained throughout the 24-week study period (Figure 5). The proportion of patients in whom the number of CD19-positive cells had recovered to at least LLN (80 cells/μl) or the baseline value, whichever was lower, by Week 24 was 80.6% in the placebo group and 6.9% in the OCR 50 mg, 3.4% in the OCR 200 mg, and 0% in the OCR 500 mg groups.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Median peripheral blood CD19-positive B cell counts over time. The lower limit of normal (LLN) was 80 cells/μl. The patient numbers shown with each investigational group represent the number of patients in that group at Time 0. Numbers shown above the different timepoints represent total number of patients in the study at that timepoint. OCR: ocrelizumab.
DISCUSSION
Our double-blind placebo-controlled study demonstrated the safety profile of OCR in Japanese patients with RA. The OCR clinical development program in patients with RA was terminated because the risk of SI outweighed the clinical benefits observed in patients with RA, based on the data from our trial and multinational clinical trials of OCR.
In our study, the majority of AE were IRR and infections, and the incidence of IRR was consistent with results reported for anti-CD20 antibodies7,8,12. Characteristic IRR symptoms in the OCR group were hypertension in 7 patients (6.1%), headache in 5 (4.4%), pyrexia in 4 (3.5%), and pruritus in 4 (3.5%); these results did not differ from previous studies of OCR or RTX.
By Week 24, SI had occurred only in the OCR group. In the OCR groups combined, the 7 patients who developed SI and the 107 patients who did not develop SI had comparable baseline white blood cell (WBC), neutrophil, and lymphocyte counts and immunoglobulin (IgG, IgM, and IgA) levels. The WBC, neutrophil, and immunoglobulin levels did not fall below LLN [WBC < 3900/μl; neutrophils < 1500/μl; IgG < 870 mg/dl; IgM < 33 mg/dl (males), < 46 mg/dl (females); IgA < 10 mg/dl] in any of the 7 patients with SI during our study, but the lymphocyte count did fall below 500/μl during the study period in 2 patients with SI. In the OCR groups combined, 2 of the 9 patients (22%) whose lymphocyte counts fell below 500/μl developed SI, while 5 of the 105 patients (4.8%) with lymphocyte count > 500/μl developed SI. Among the SI, PCP occurred in 2 patients in the OCR 200 mg group. At the onset of PCP, both patients exhibited pyrexia, hypoxemia, pulmonary ground-glass opacity, and increased serum ß-D-glucan levels, and 1 patient was positive on the polymerase chain reaction test for P. jirovecii. Both patients recovered with methylprednisolone pulse therapy and trimethoprim-sulfamethoxazole. Advanced age, concurrent lung disease, and concomitant corticosteroid use have been reported as risk factors for bacterial pneumonia or SI including PCP during treatment of Japanese RA patients with tumor necrosis factor (TNF) inhibitors17,18,19,20,21. In our study, 6 of the 7 patients with SI were using prednisolone concomitantly, but only 3 of the 7 patients with SI were over 60 years of age (3 were in their 40s and 1 in her 20s) and none had concurrent lung disease. Similarly, both patients who developed PCP in our study were taking 7.5 mg/day oral prednisolone, but both were 42 years old and did not have concurrent lung disease. Further, their lymphocyte counts at onset of PCP were decreased to 651 and 890/μl, respectively. These results suggest that risk factors for SI, including PCP, during OCR treatment may differ from those during treatment with TNF inhibitors.
Although there were differences of sample size, patient background, and observation period, the incidence of SI in our study (6.1%) was comparable to the results of other Japanese clinical trials of biologic agents: 7.6% in a 52-week study of tocilizumab in patients with inadequate response to DMARD (SAMURAI study)22; 3.3% in a 24-week study of tocilizumab in patients with inadequate response to MTX (SATORI)23; 5.2% in a 54-week study of infliximab in patients with inadequate response to MTX (RISING)24; and 4.9% in a 24-week study of adalimumab in patients with inadequate response to DMARD (CHANGE)25. PCP was observed in 2 patients (1.75%) in our study, but in the SAMURAI, SATORI, and CHANGE studies, no PCP was observed. Further, the incidence of PCP in our study was higher than those in the Japanese postmarketing surveillance data of biologic agents (tocilizumab, infliximab, and etanercept), that is, 0.2% to 0.4%17,18,19,26. These results suggest that treatment with OCR in Japanese patients with RA may have higher risk for PCP than treatment with the other biologic agents.
A possible association of efficacy with B cell depletion was reported in patients with RA treated with OCR in the ACTION study12, which ascertained that B cell depletion was maintained until Week 24 in groups that received ≥ 200 mg OCR, but was not maintained in groups with lower dosages. Significant improvements of signs and symptoms of RA shown by relatively stringent response criteria, including ACR70 response, DAS28-ESR clinical remission, and EULAR good response, were also obtained only in groups that received ≥ 200 mg OCR12. The clinical response to RTX has been reported to be determined by the level of B cell depletion rather than by the dose of the drug27. In our study, the percentages of patients with peripheral blood B cell count at Week 24 that was above LLN or baseline values were 6.9%, 3.4%, and 0% in the OCR 50, 200, and 500 mg groups, respectively. The OCR 200 mg group showed higher clinical responses than the other 2 OCR groups in every efficacy criterion used in our study. In addition, the peripheral B cell count recovered to at least the LLN (80 cells/μl) or the baseline value in 4 patients in the OCR groups combined, but these patients showed sustained efficacy through Week 24. It is difficult to draw firm conclusions because of the small number of patients with B cell recovery and the limited study period (24 weeks), but these results suggest that peripheral B cell count alone may not account for maintenance of efficacy in patients with RA treated with OCR.
As a limitation of our study, we note the dosage of MTX. The mean MTX dosage in each group was 7.3–7.6 mg/week, which was lower compared to clinical trials of OCR for RA conducted in some Western countries. The approved maximum dose of MTX was 8 mg/week in Japan when this trial was implemented and we had to design the trial under this restriction. This should be taken into account when interpreting our results.
Serious infections, including PCP, occurred only in the combined OCR groups in our study, possibly indicating an elevated risk for SI from OCR use in Japanese patients with RA. Treatment with OCR resulted in better clinical responses than treatment with the placebo in Japanese RA patients with an inadequate response to MTX, about 30% of whom had been previously treated with a biological DMARD. Although we should take into account the small sample size and the premature termination of the study, these results would suggest an inappropriate benefit-risk balance for OCR in this patient population. Because of the lack of approval for RTX for RA in Japan and the recommended use of the drug for patients with RA who have failed TNF inhibitor therapy in Western countries, it will be necessary to investigate the safety and efficacy of other anti-B cell therapies in Japanese patients with RA.
Acknowledgment
The authors thank the patients who participated in the study and the investigators of the JA21963 Study Group.
APPENDIX
List of study collaborators. Primary investigators of the JA21963 study group: Kazuhide Tanimura (Hokkaido Medical Center for Rheumatic Diseases), Hiroki Takahashi (Sapporo Medical University), Yukitomo Urata (Seihoku Central Hospital), Yasuhiko Hirabayashi (Hikarigaoka Spellman Hospital), Tomonori Ishii, Hiroshi Fujii (Tohoku University Hospital), Takayuki Sumida (Tsukuba University Hospital), Chihiro Terai (Jichi Medical University Saitama Medical Center), Ryutaro Matsumura (National Hospital Organization Chiba-East Hospital), Makoto Sueishi (National Hospital Organization Shimoshizu Hospital), Kazuhiko Yamamoto (The University of Tokyo Hospital), Akio Yamada, Daitaro Kurosaka (Jikei University School of Medicine), Akio Mimori (International Medical Center of Japan), Yusuke Miwa (Showa University Hospital), Masataka Kuwana (Keio University Hospital), Shinichi Kawai (Toho University Omori Medical Center), Yoshiaki Ishigatsubo (Yokohama City University Hospital), Kazunori Sugimoto (Fukui General Clinic), Noriyoshi Ogawa (Hamamatsu University School of Medicine), Toshiaki Miyamoto (Seirei Hamamatsu General Hospital), Shigenori Tamaki, Motokazu Kai (National Hospital Organization Mie Chuou Medical Center), Daisuke Kawabata (Kyoto University Hospital), Toshio Tanaka (Osaka University Hospital), Masaaki Inaba (Osaka City University Hospital), Shunichi Kumagai, Akio Morinobu, Yasushi Miura (Kobe University Hospital), Hajime Sano (Hyogo College of Medicine), Naoki Kashihara, Yoshitaka Morita (Kawasaki Medical School Hospital), Kazuhiko Ezawa (Kurashiki Kosai Hospital), Yuji Yamanishi, Masanori Kawashima (Hiroshima City Hospital), Seizo Yamana, Mitsuhiro Iwahashi (Higashihiroshima Memorial Hospital), Hiroaki Dobashi (Kagawa University), Kiyoshi Takasugi (Dohgo Spa Hospital), Takahiko Horiuchi (Kyusyu University Hospital), Eiichi Suematsu (National Hospital Organization Kyushu Medical Center), Takaaki Fukuda (Kurume University Medical Center), Katsumi Eguchi, Atsushi Kawakami (Nagasaki University Hospital).
Footnotes
-
Supported by Chugai Pharmaceutical Co. Ltd. M. Harigai has received research grants, consultant fees, and/or speakers’ bureau honoraria from Abbott Japan, Bristol-Myers Japan, Chugai Pharmaceutical Co. Ltd., Eisai Co. Ltd., Janssen Pharmaceutical KK, Mitsubishi Tanabe Pharma, Pfizer Japan Inc., and Takeda Pharmaceutical Co. Ltd. Y. Tanaka has received consulting fees, speaking fees, and/or honoraria from Chugai, Mitsubishi-Tanabe, Eisai, Takeda, Astellas, and Abbott and has received research grant support from Chugai, Mitsubishi-Tanabe, Takeda, MSD, Pfizer, Astellas, Abbott, and Eisai.
-
Full Release Article. For details see Reprints/Permissions at jrheum.org
- Accepted for publication October 13, 2011.
Free online via JRheum Full Release option
REFERENCES
- 1.
- 2.
- 3.
- 4.
- 5.
- 6.
- 7.
- 8.
- 9.
- 10.
- 11.
- 12.
- 13.
- 14.
- 15.
- 16.
- 17.
- 18.
- 19.
- 20.
- 21.
- 22.
- 23.
- 24.
- 25.
- 26.
- 27.