
To the Editor:
Combined pulmonary fibrosis and emphysema (CPFE) has recently been defined as a distinct syndrome occurring in adult heavy smokers in their sixties and characterized by dyspnea, crackles at lung auscultation, and diagnostic imaging features on high resolution computed tomography (HRCT) of the chest1. Spirometry is often normal or subnormal due to the opposite compensatory effects of pulmonary fibrosis (usually resulting in restrictive lung disease) and of emphysema (usually resulting in obstructive lung disease)1.
CPFE has been reported in a series of patients with connective tissue disease (CTD) as a novel hitherto unrecognized pulmonary manifestation2. We describe a “radical” case of CPFE in a young woman with severe systemic sclerosis (SSc) yet with no history of heavy smoking.
A 28-year-old woman, an ex-smoker of tobacco (less than 5 pack-years over 10 years) and ex-smoker of cannabis (1 joint per week for 5 years), presented with mild dyspnea. She had no significant environmental or occupational exposure. Limited cutaneous SSc was diagnosed at the age of 15 years, including sclerodactyly, Raynaud phenomenon, reduction of the oral aperture, scars from ulcerations at the fingertips (treated with bosentan orally for 5 years), and gastroesophageal reflux. She denied any joint or muscle symptoms. Bilateral basal crackles (“velcro rales”) were present at lung auscultation.
Spirometry was normal: forced vital capacity was 99% of predicted value; total lung capacity, 108%; forced expiratory volume in 1 s, 75%; forced expiratory volume in 1 s/vital capacity, 65% (78% of predicted value). However, further pulmonary function tests disclosed inflation with residual volume of 126% of predicted; residual volume/total lung capacity, 33% (116% of predicted value) and impairment of gas exchange with single-breath carbon monoxide diffusion factor 47% of predicted; and PaO2 at rest 13.8 kPa decreasing to 10.2 kPa after 10-min exercise of 50 W. Body mass index was 20.5 kg/m2.
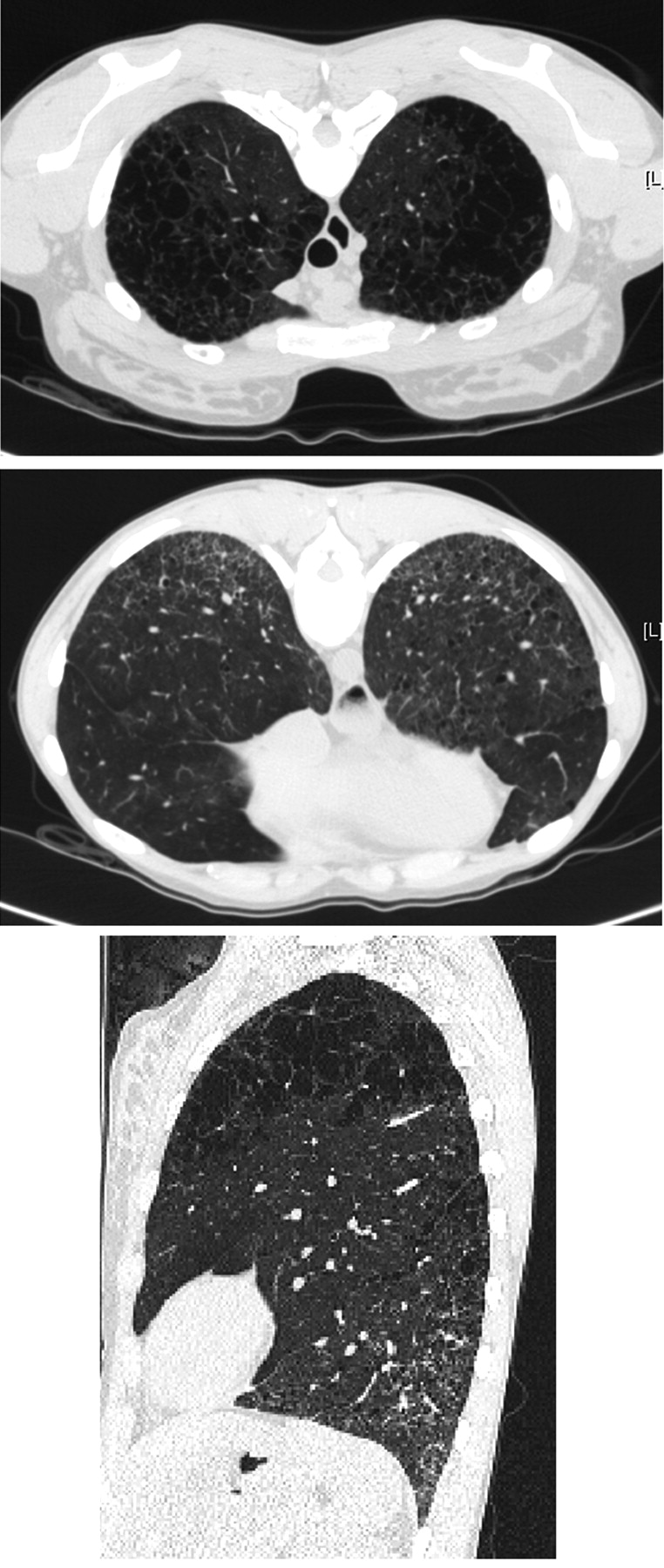
HRCT of the chest showed severe centrilobular emphysema in the upper zones of the lungs, and diffuse infiltrative lung disease in the lower zones with predominant ground-glass attenuation, bilateral reticulation without subpleural predominance, and traction bronchiectasis (Figure 1). The bronchoalveolar lavage differential cell count was normal: 86% macrophages, 10% lymphocytes, and 4% neutrophils. Echocardiography was normal, with estimated systolic pulmonary arterial pressure 30 mm Hg. Antinuclear antibodies with a heterogenous pattern and high titer (1/5120) were further characterized as anti-U1-RNP autoantibodies. Alpha-1 antitrypsin level and phenotype were normal. No systemic therapy was initiated.

{kind=link}
Computed tomography scan reveals centrilobular emphysema in the upper zones of the lungs (upper panel), reticulation and architectural distortion of the lung bases (middle panel), and 2-dimensional reconstruction (lower panel).
This unconventional case of CPFE syndrome in a patient with SSc is unique by its occurrence in a young patient with only mild smoking history. Mean age at diagnosis was between 65 and 70 years in the largest series of “idiopathic” CPFE1,3,4,5,6, and no patient was previously diagnosed before the age of 36 years1.
CPFE syndrome is characterized by unexpected subnormal or normal spirometry contrasting with severely impaired diffusion capacity, hypoxemia at exercise, and frequent pulmonary hypertension3. Diagnosis relies on chest HRCT that typically shows centrilobular and/or paraseptal emphysema, and diffuse interstitial opacities suggestive of pulmonary fibrosis of the lower lobes1. The chest imaging in our patient was typical of CPFE syndrome with both conspicuous centrilobular emphysema in the upper lobes and a pattern suggestive of nonspecific interstitial pneumonia in the lung bases.
This syndrome is mainly related to heavy tobacco smoking, with about 95% of smokers and a mean of ∼40 pack-years in CPFE patients without CTD1,3,4,5,6. CPFE has been reported recently in patients with CTD, especially rheumatoid arthritis and SSc2. The occurrence of CPFE in never-smokers with CTD is intriguing2. Smoking history less than 5 pack-years in our patient suggests that the CTD played a role in the development of CPFE lesions. Interestingly, emphysema is more prevalent in SSc patients with pulmonary fibrosis than in control smokers with CTD or idiopathic pulmonary fibrosis, after adjustment for the smoking history8. Spontaneous emphysema also develops in tight-skin mice9 that resembles human SSc. This observation thus indicates that risk factors or etiologies other than tobacco smoking may be involved in CPFE, including CTD. The report of a mutation in the surfactant protein-C gene in a young nonsmoking woman with CPFE7 further supports the hypothesis of a genetic predisposition in some patients with CPFE.
CPFE causes significant morbidity, as overall survival was estimated at 73% at 5 years in CPFE and CTD, in which 5 of 10 patients with SSc developed pulmonary arterial hypertension2, the main determinant of survival in CPFE1.
Our observations provide novel information that in SSc the CPFE syndrome may occur in a young adult with only mild smoking history.
REFERENCES
- 1.
- 2.
- 3.
- 4.
- 5.
- 6.
- 7.
- 8.
- 9.