
Abstract
Objective. Naproxcinod, a cyclooxygenase-inhibiting nitric oxide donator antiinflammatory drug, was evaluated in this phase 2, double-blind, randomized, parallel group study to determine its optimal dose in patients with osteoarthritis (OA).
Methods. In total 543 patients with OA of the hip or knee were randomized to receive naproxcinod 750 mg once daily (qd), 750 mg twice daily (bid), 1125 mg bid, rofecoxib 25 mg qd, or placebo for 6 weeks. The primary efficacy variable was the within-patient change from baseline to the average of Weeks 4 and 6 in WOMAC™ pain subscale score. Treatment-group differences were compared using ANCOVA with factors for treatment and country, and baseline pain subscale score as a covariate. Safety endpoints included vital signs and adverse events. Treatment-group differences in mean change from baseline to Week 6 in systolic blood pressure (SBP) were compared using an ANCOVA with treatment and country as fixed factors and baseline SBP as covariate.
Results. All active treatments showed statistically significant reductions in WOMAC pain score compared to placebo (p ≤ 0.02). Naproxcinod was well tolerated. The 750 mg bid dose appeared to have the best balance of benefit versus safety. All 3 naproxcinod doses showed a reduction in SBP, while an increase was shown for rofecoxib. The changes for the naproxcinod groups were statistically significantly better compared to rofecoxib (p ≤ 0.02).
Conclusion. This dose-finding study identified naproxcinod 750 mg bid as the upper dose for further therapeutic confirmatory clinical trials. Naproxcinod at all doses decreased mean SBP compared to an increase with rofecoxib.
- NAPROXCINOD
- OSTEOARTHRITIS
- CYCLOOXYGENASE-INHIBITING NITRIC OXIDE DONATOR
- ROFECOXIB
Osteoarthritis (OA) is the leading cause of physical disability in industrialized nations1,2. The socioeconomic effect of the disease is great, with approximately 43 million patients in the US and an annual cost to society of roughly $65 billion. This burden is expected to increase by 25% by 2020, partly due to the aging population and a higher incidence of obesity3,4.
Nonsteroidal antiinflammatory drugs (NSAID) are widely used to treat OA. However, cyclooxygenase-2 (COX-2) inhibitors and traditional NSAID tend to cause adverse effects on the cardiovascular (CV) system, including eliciting and exacerbating hypertension, precipitating congestive heart failure and renal insufficiency, and increasing the risk of thrombotic events5–7. Thus, new and potentially safer drugs are needed for successful management of OA.
Naproxcinod is the most advanced of the cyclooxygenase-inhibiting nitric oxide (NO) donator (CINOD) class of antiinflammatory drugs designed for treatment of chronic pain. Upon absorption, naproxcinod is rapidly cleaved to produce naproxen and a NO-donating moiety. Naproxen results in inhibition of COX-1 and COX-2 and is known to be an effective antiinflammatory and analgesic agent. The release of NO results in effects on the CV system as well as protection of the gastrointestinal (GI) tract and other organs8,9. NO possesses marked vascular smooth-muscle relaxant properties through activation of soluble guanylyl cyclase and consequent formation of cyclic guanosine monophosphate (cGMP)8. As such, NO continuously generated in vascular endothelium is critical for homeostasis of the vascular tone. It has been shown that impairment in NO vascular activity is a contributing factor in the pathogenesis of hypertension10. Exogenous NO administration leads to vasorelaxation and decreases elevated blood pressure (BP) in a variety of experimental models and clinical conditions11–13. CINOD have been designed to overcome the side effects arising from chronic administration of NSAID, exploiting the activity of NO on the CV and GI systems.
Our study was designed to assess the efficacy of different doses and dosing regimens of naproxcinod in relation to placebo as well as the active comparator, rofecoxib, in patients with OA of the knee or hip. Additionally, the overall safety of naproxcinod was evaluated with specific attention to the effects of naproxcinod on BP.
MATERIALS AND METHODS
This randomized double-blind placebo-controlled parallel-group dose-finding study, conducted at 61 centers in Europe, Australia, and the USA, compared the analgesic efficacy, safety, and tolerability of 3 dosing regimens of naproxcinod with rofecoxib and placebo in patients with OA of the hip or knee.
The study was performed in accord with the Declaration of Helsinki and all patients provided written informed consent prior to participating in study procedures. Each study center obtained local ethics committee/institutional review board approval prior to commencement.
Eligibility criteria
We studied male and female patients aged ≥ 40 years with primary OA of the hip or knee that had necessitated use of NSAID for a minimum of 15 days during the 4 weeks prior to the screening visit, satisfying the American College of Rheumatology (ACR) classification criteria for diagnosis of OA in the target joint, with diagnoses confirmed by radiography. Patients were also required to have ACR global functional status class I, II, or III14. At screening, eligible patients had to experience pain in the target joint during loading, recorded as ≤ 80 mm on a 100-mm visual analog scale (VAS) of the first question of the Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC™) pain subscale (version 3.1)15–17. Patients were also required to experience a pain flare within 3–14 days of discontinuing all pain medications during a washout phase (between screening and baseline). The VAS pain score for pain on walking on a flat surface at baseline was required to be ≥ 40 mm, with an increase of at least 15 mm compared to screening.
Exclusion criteria included secondary OA, a diagnosis of arthritis other than OA, a hip or knee replacement in the target joint, history of gastric or duodenal ulceration within 3 months or gastroduodenal bleeding within 6 months of screening, supine systolic blood pressure (SBP) < 110 mm Hg, NSAID hypersensitivity, renal impairment, treatment with > 325 mg per day of aspirin or opioids within 48 hours prior to screening or > 4000 mg per day of acetaminophen (paracetamol). Acetaminophen ≤ 4000 mg/day could be taken as a rescue medication during the study for patients who experienced unacceptable pain for more than 24 hours. Antihypertensive drugs were allowed if the dose had been stable for 3 months prior to the screening visit.
Randomization and blinding
Eligible patients were sequentially randomized at baseline, using a computer-generated list, to receive naproxcinod 750 mg once daily (qd), naproxcinod 750 mg twice daily (bid), naproxcinod 1125 mg bid, rofecoxib 25 mg qd, or placebo for 6 weeks. The patient numbers were assigned to centers in blocks, and the investigators were blinded to the treatment allocation. To ensure masking, all treatments were provided in identical capsules (using an overencapsulated form of rofecoxib) and patients receiving naproxcinod 750 mg qd and rofecoxib 25 mg qd also received a matching placebo capsule in the evening. A patient diary was used to record dosing details of the study treatment, rescue analgesia, and other concomitant medications used during the treatment period.
Efficacy and safety evaluations
The primary efficacy variable was the within-patient change from baseline to the average score of Weeks 4 and 6 in WOMAC pain subscale score. Secondary endpoints included changes in disease-specific symptoms according to subscale scores of pain, stiffness, and function of the WOMAC, daily VAS pain intensity at rest and during walking, patient and investigator rating of disease status, response to therapy and overall rating of treatment, and the average weekly amount of rescue medication taken by each patient. Patients were monitored for clinical or laboratory adverse events by physical examination, vital signs, electrocardiography, hematology, blood chemistry, and urinalysis. Pulse and BP measurements were taken at Visit 1 (screening), Visit 2 (baseline), Visit 3 (Week 1), Visit 4 (Week 2), Visit 6 (Week 4), Visit 8 (Week 6), and Visit 9 (followup). Office BP monitoring was performed after 15-minute rest with a calibrated manometer in both the supine and standing position. At each visit, supine measurements were made using the same arm, where the highest BP was recorded at Visit 1. Where possible, the same person performed all measurements on the same subject. Orthostatic measurements were performed at 2 and 5 minutes when going from a supine to a standing position.
Statistical analysis
No formal sample size calculations were performed since this study was considered exploratory in design. However, as a measure of study precision, the maximum half-length of the 95% confidence intervals (CI) expected with at least 80% coverage probability for treatment group differences in WOMAC pain subscale (mm) was ± 4.4 within one active treatment, ± 6.1 between 2 active groups, and ± 6.9 between active and placebo groups. These CI were not adjusted for statistical significance. As a measure of assay sensitivity, the planned number of evaluable patients was to provide at least 90% power to detect a difference ≥ 11 mm between the rofecoxib and placebo treatment groups in the WOMAC pain subscale score. This assumed a 2-sided t test, 5% level of significance, and standard deviation of 20 mm.
The primary efficacy analysis was based on the full analysis set comprising all patients who received at least one dose of investigational product and provided a baseline and at least one post-baseline WOMAC pain subscale score. The full analysis set was analyzed according to the study drug actually received. A per-protocol analysis set was defined as a subset of the full analysis set including only patients who completed the study without major protocol deviations.
The “last available value carried forward” principle was used to determine missing values. Only post-baseline values were carried forward. Treatment-group differences were compared using analysis of covariance (ANCOVA), with factors for treatment-group and country grouping and a covariate for the baseline pain subscale score. No adjustments for multiple comparisons were made due to the exploratory design of the study.
The safety analysis included all patients who took at least one dose of the investigational product. Safety variables were analyzed primarily by means of descriptive statistics according to the treatment the patient actually received. Treatment group differences in mean changes of SBP from baseline at Week 6 were analyzed post-hoc, using an ANCOVA model, with treatment and country grouping as fixed factors and baseline SBP as covariate.
RESULTS
Patient disposition
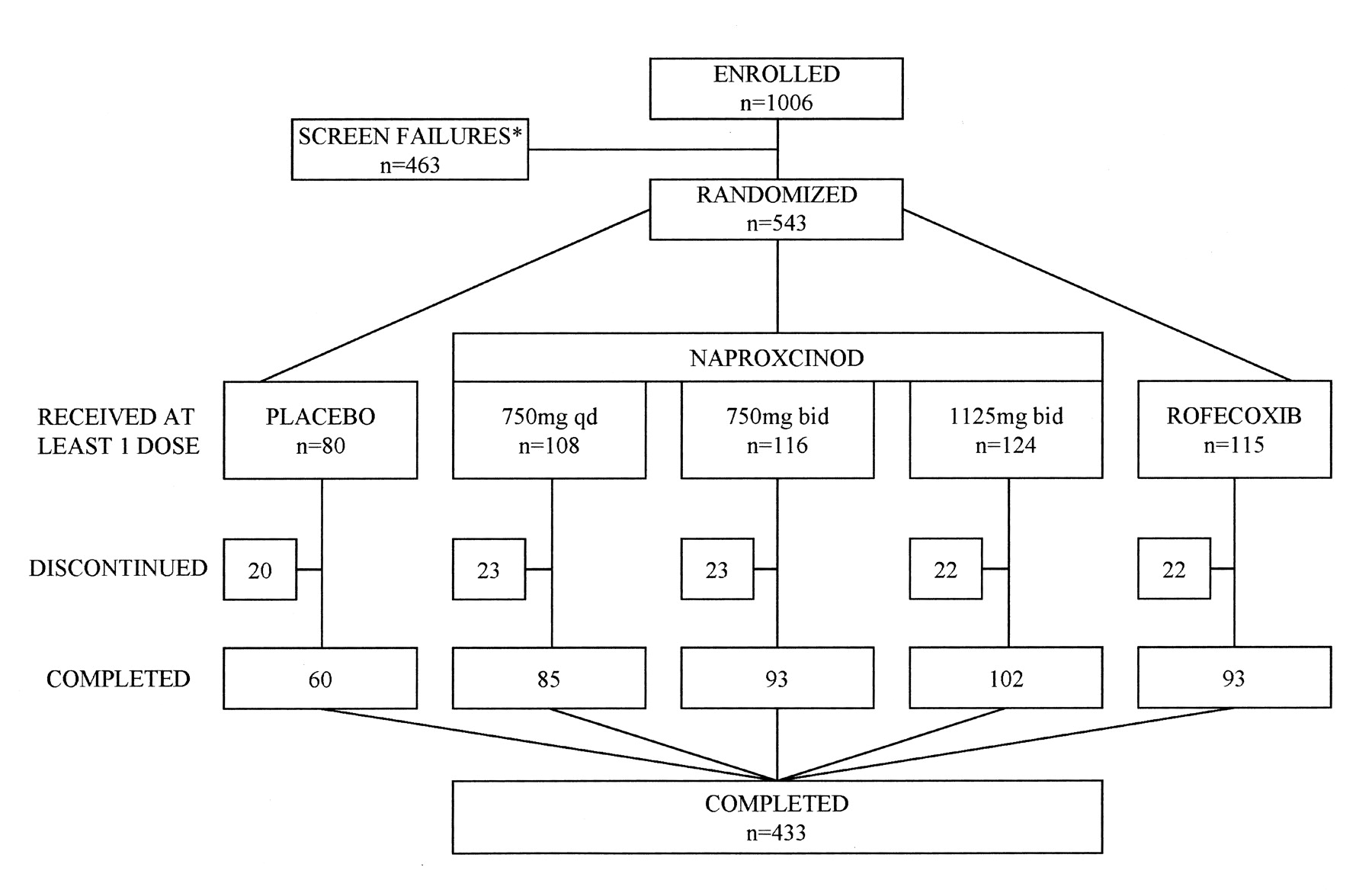
A total of 1006 patients were screened, with 543 patients randomized to treatment. All randomized patients were analyzed for safety, 522 were analyzed for efficacy in the full analysis set, and 404 were analyzed in the per-protocol set. Overall, 433 patients (80%) completed the study (Figure 1). In total, 110 (20%) subjects discontinued from study drug; these were evenly balanced across the treatment groups, with the exception of the placebo group, where a higher proportion discontinued due to lack of efficacy (10% compared to a range of 3% to 7% in the active treatment groups), and adverse events for rofecoxib. The treatment groups were comparable for demographic and baseline characteristics and were representative of the overall OA population (Table 1).

Patient disposition: all-patients analysis set. * Ineligible 88%; consent withdrawn 12%.
Demographic data and baseline characteristics of the full analysis set of subjects.
In the safety population, the lowest proportion of patients taking antihypertensive medications prior to the intake of study drug was in the placebo group (29%), while all other groups were similar, ranging between 36% (naproxcinod 1125 mg bid group) and 44% (naproxcinod 750 mg qd group). The intake of gastroprotective agents prior to randomization was balanced across treatment groups (range 12% to 18%).
Efficacy
The primary efficacy variable was the within-patient difference between the WOMAC pain subscale score at baseline and the average of Weeks 4 and 6 (Table 2).
WOMAC pain score (mm) at baseline, mean change from baseline to the average of Weeks 4 and 6, and differences between treatment groups of the full analysis set. Calculated using ANCOVA with treatment, country grouping (Australia, USA, Europe) and baseline pain as covariates.
In the full analysis set, naproxcinod 750 mg qd, 750 mg bid, 1125 mg bid, and rofecoxib 25 mg qd were statistically superior to placebo in reducing the WOMAC pain subscale score from baseline to the average of Weeks 4 and 6. Naproxcinod 750 mg bid, naproxcinod 1125 mg bid, and rofecoxib 25 mg qd were not statistically significantly different from each other, but were all superior to naproxcinod 750 mg qd. Results for the per-protocol set generally reflected those of the full analysis set.
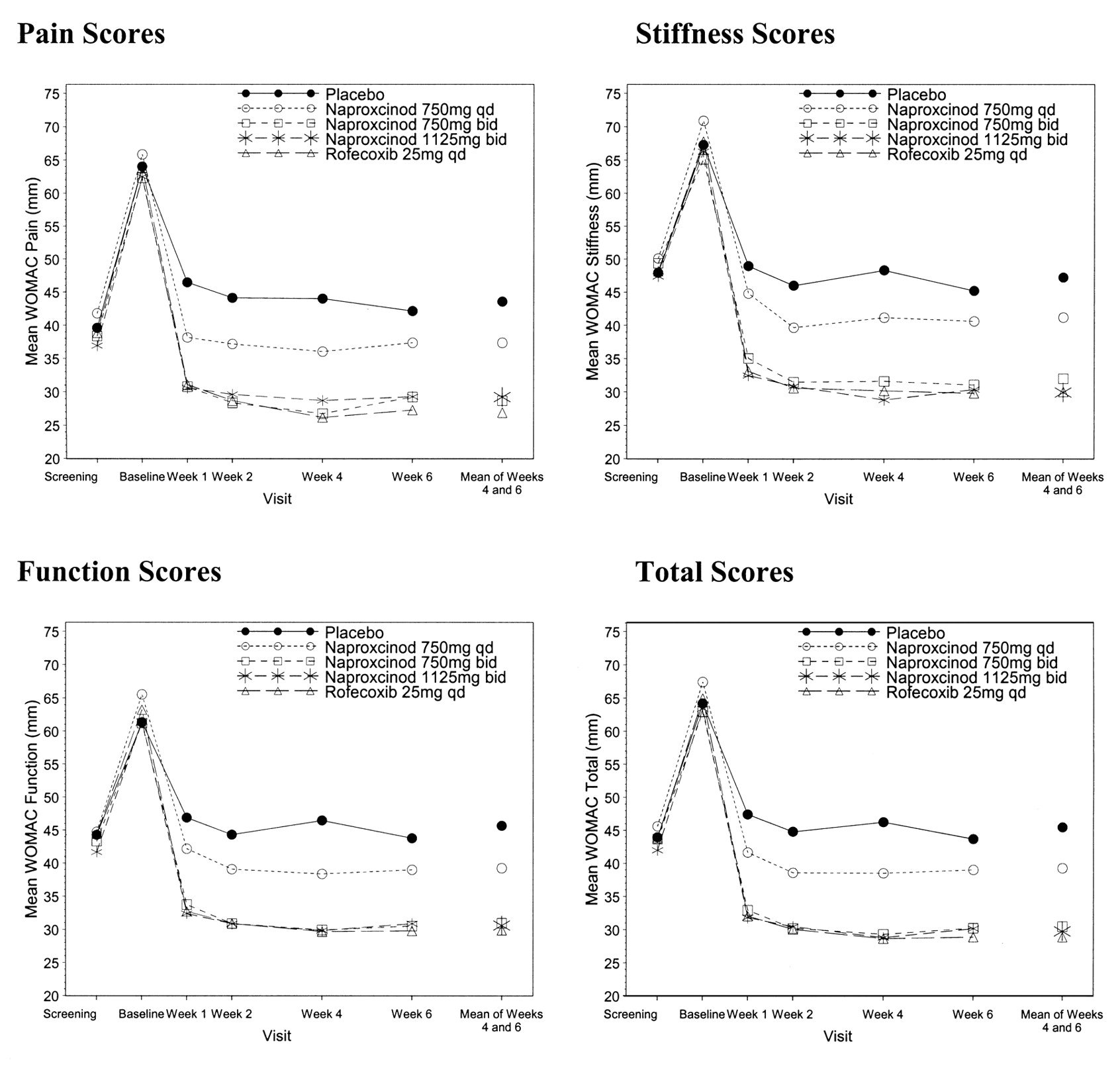
Secondary variables of WOMAC pain, stiffness, function, and total scores, and VAS pain intensity supported the findings of the primary variable, indicating that all active treatment groups were significantly better than placebo (Figure 2). Patients’ and investigators’ ratings of treatment, disease status, response to therapy, and rescue medication usage indicated that all active treatment groups were statistically significantly better than placebo.

{kind=link}
{kind=link}
WOMAC pain, stiffness, function, and total scores over time: full analysis set (imputed for last available value carried forward). BL: baseline; W: week. S: screening
Safety and tolerability
The majority of patients in the safety analysis set achieved ≥ 90% compliance (median compliance 99% to 100%), and were exposed to study drug for between 36 and 49 days (median exposure 42 to 43 days). This was balanced across the treatment groups. Taken together, these high compliance and duration of exposure values give confidence that the patients’ exposure to study drug was adequate for the evaluation of safety.
Overall, naproxcinod was generally well tolerated. Five patients experienced a total of 6 serious adverse events, most of them in the placebo group (4 serious adverse events in 3 patients). Only one serious adverse event was considered drug-related by the investigator (an abnormal electrocardiogram T-wave in a patient taking naproxcinod 1125 mg bid). No serious adverse events were reported in naproxcinod 750 mg qd or bid groups.
Forty-two patients discontinued permanently from study drug due to adverse events. Discontinuations due to adverse events were reported in all treatment groups, but were proportionally higher in the rofecoxib 25 mg qd group (10% compared to a range of 6% to 8% in the other active treatment groups and 9% in the placebo group).
The most commonly reported adverse events were headache, dyspepsia, nausea, and dizziness, the latter being slightly higher in the naproxcinod groups but showing no relationship to dose. GI disorders were reported somewhat more frequently with the higher doses of naproxcinod, although evaluation of upper GI-related symptoms (dyspepsia, nausea, and upper abdominal pain) failed to show a dose-response relationship (Table 3).
Number (%) of patients with the most commonly reported adverse events (AE) (≥ 5% in at least one treatment group) during the treatment period in the safety analysis set.
All 3 naproxcinod dose groups showed a reduction in SBP from baseline to Week 6 (pre-dose), while an increase in SBP was seen in the rofecoxib group (Table 4). The baseline pre-dose SBP values were similar in all groups, ranging from 135 to 139 mm Hg. There was a statistically significant difference in favor of all 3 naproxcinod doses as compared to rofecoxib in the change from baseline to Week 6 (p = 0.0131 for naproxcinod 750 mg qd; p = 0.0044 for naproxcinod 750 mg bid; p < 0.0001 for naproxcinod 1125 mg bid). No statistically significant differences were seen for comparisons of naproxcinod versus placebo. At Week 6, the percentage of patients with a SBP increase ≥ 3 mm Hg was 25% for placebo, 48% for rofecoxib, and 27%, 28% and 20% for naproxcinod 750 mg qd, 750 mg bid, and 1125 mg bid, respectively. The percentage of patients that showed a decreased SBP of ≤ 3 mm Hg was 46% for placebo, 23% for rofecoxib, 43% for both 750 mg naproxcinod groups, and 52% for naproxcinod 1125 mg bid. There were no major differences, over the course of the study, between treatment groups in the proportion of subjects with treatment-emergent decreases of > 30 mm Hg in supine SBP. A slightly higher proportion of subjects in the naproxcinod groups had treatment-emergent decreases > 15 mm Hg in SBP during the study.
Baseline, Week 6, and change from baseline at Week 6 in mean supine systolic blood pressure (mm Hg) in the safety analysis set.
DISCUSSION
Despite the range of options available to treat OA, there remains a need for therapies that offer either improved efficacy or enhanced safety. Our study of naproxcinod was directed at investigating both these potential benefits as well as attempting to establish an optimal dosing interval.
NSAID are the drugs most widely used for the treatment of OA. Unlike opioids, which have a wide dynamic range for pain relief, NSAID demonstrate a ceiling effect for pain relief with increasing dose18–21. This property is clinically important as the overall safety of NSAID appears to be inversely related to dose, underscoring the importance of being able to identify the optimal dosing regimen that will result in maximal efficacy with minimal side effects. Increasing doses above this level is not likely to result in increased pain relief, but can lead to higher incidence of serious side effects.
In this study, naproxcinod at 2 different doses, 750 mg and 1125 mg, was evaluated versus placebo and rofecoxib 25 mg daily, with the 750 mg dose evaluated at a once-daily and a twice-daily dosing regimen. A flare design, a standard methodology in efficacy trials of NSAID, was utilized in contrast to the approach used in clinical practice. Earlier studies in OA of the knee had indicated that naproxcinod 375 mg and 750 mg twice daily were both efficacious, the higher dose showing greater efficacy22,23. In this study, both the 750 mg and 1125 mg bid doses of naproxcinod were clinically efficacious, demonstrating a decrease in the WOMAC pain subscale score of 13–14 mm compared to placebo, and similar to that of rofecoxib 25 mg qd. However, naproxcinod 1125 mg bid failed to show any enhanced efficacy compared to the 750 mg bid dose in any of the standard outcome measures evaluated. Thus, these data permit definition of the upper end of the dose-response curve of efficacy for naproxcinod. In terms of dosing frequency, once-daily dosing of 750 mg naproxcinod was also assessed and was found to have efficacy greater than placebo, but pain relief and functional change were statistically significantly less than with the twice-daily dosing regimen. This result is consistent with the known half-life of the drug24.
Providing improved safety is a second approach to delivering new beneficial therapeutic options. Of the important mechanism-based side effects of NSAID, GI and CV toxicity are arguably the most prevalent. Naproxcinod offers the advantage, in addition to naproxen, of liberating a NO-donating moiety. NO is known to have both physiologic and pharmacologic properties that could prove advantageous in the setting of cyclooxygenase inhibition. In accord with findings in animals25,26, clinical studies in humans showed that naproxcinod may account for a reduction of erosions and ulcers in the stomach and duodenum with respect to naproxen23,27,28.
In vascular smooth muscle cells, NO acts almost exclusively via the enzyme soluble guanylate cyclase, producing vasodilatation and BP modulation via cGMP-dependent protein kinases29. NO is also known to have many other beneficial effects on the vascular endothelium, including inhibition of vascular smooth muscle cell proliferation, an inhibitory effect on platelet aggregation and adhesion, and inhibition of inflammatory cell activation30–32. A differentiated BP profile of naproxcinod compared to naproxen and rofecoxib has been shown in a previous phase 2 study22.
Evaluation of general safety in this study demonstrated similar findings among all active treatment arms. With GI-related adverse events, there was little difference between any of the naproxcinod arms and rofecoxib when considering upper GI symptoms (dyspepsia, nausea, upper abdominal pain), with all groups having a slightly higher incidence than reported for placebo, although the numbers in each group were limited. Demonstration of the effects of NO on GI outcomes, either symptoms or ulcers, will require larger appropriately powered studies.
Particular attention was paid in this study to the BP response to treatment, as BP is known to be affected by NSAID. Rofecoxib treatment, as demonstrated, resulted in a statistically significant increase in BP compared to placebo33,34. In contrast, naproxcinod, in each of the treatment groups, resulted in a statistically significantly lower BP than rofecoxib, and demonstrated no significant difference compared to placebo. More precise definition of comparative BP response to naproxcinod and NSAID will require larger studies with additional comparators. There were no reports of hypotension in this study. The prevalence of dizziness appeared slightly higher in the naproxcinod groups compared to the other treatment arms, although differences were small and no dose-response relationship was seen.
The results of this dose-finding study support the conclusion that there is no additional efficacy of naproxcinod at doses higher than 750 mg bid, and that this dose is clinically effective and similar to that seen with rofecoxib 25 mg qd. Additionally, the safety data from this phase 2 study support the differentiation of naproxcinod from other NSAID in terms of its effect on BP. Since this was a small study whose primary focus was not on BP differentiation, additional studies are needed to better define the advantages NO release may have on BP maintenance in patients with OA. However, as hypertension is present in approximately 50% of this population, and BP destabilization is a problem with NSAID therapy, the potential of ameliorating this side effect warrants further scrutiny.
This dose-finding study identified naproxcinod 750 mg bid as the upper dose for further therapeutic confirmatory clinical trials. Naproxcinod 375 mg bid was identified as the lowest efficacious dose in a previous trial22. Both trials showed that naproxcinod at all doses decreased mean SBP compared with rofecoxib. Therefore both naproxcinod 375 mg and 750 mg bid are under investigation in larger trials over longer periods of time to define potential use in management of OA.
Acknowledgments
Medical writing support was provided by Debbie Jordan.
Footnotes
-
Supported by NicOx S.A. J. Karlsson received a research grant from Astra Zeneca. T.J. Schnitzer served as consultant to Astra Zeneca, Merck, NicOx, and Novartis.
- Accepted for publication January 22, 2009.
REFERENCES
- 1.
- 2.
- 3.
- 4.
- 5.
- 6.
- 7.
- 8.
- 9.
- 10.
- 11.
- 12.
- 13.
- 14.
- 15.
- 16.
- 17.
- 18.
- 19.
- 20.
- 21.
- 22.
- 23.
- 24.
- 25.
- 26.
- 27.
- 28.
- 29.
- 30.
- 31.
- 32.
- 33.
- 34.