

Abstract
Objective To evaluate the long-term efficacy and safety of ustekinumab through 2 years in patients with active systemic lupus erythematosus (SLE).
Methods This was a placebo-controlled (week 24), phase II study in 102 patients with seropositive active SLE. Patients were randomized to ustekinumab (approximately 6 mg/kg single intravenous infusion, then subcutaneous [SC] injections of 90 mg every 8 weeks) or placebo, added to background therapy. Placebo patients initiated ustekinumab (90 mg SC every 8 weeks) at week 24. Patients could enter an optional open-label study extension after week 40 (final ustekinumab administration at week 104). Efficacy assessments included Systemic Lupus Erythematosus Disease Activity Index 2000 (SLEDAI-2K), SLEDAI-2K Responder Index-4 (SRI-4), physician global assessment (PGA), and Cutaneous Lupus Erythematosus Disease Area and Severity Index (CLASI). Observed data are reported for the extension period. The final efficacy assessment was at week 112; safety was monitored through week 120.
Results In this subset of patients who entered the study extension, 24 in the ustekinumab group and 14 in the placebo crossover group completed study treatment. At week 112, 79% and 92%, respectively, had an SRI-4 response; 92% in both groups had ≥ 4-point improvement from baseline in SLEDAI-2K score; 79% and 93%, respectively, had ≥ 30% improvement from baseline in PGA; 86% and 91%, respectively, had ≥ 50% improvement in active joint (pain and inflammation) count; and 79% and 100%, respectively, had ≥ 50% improvement in CLASI Activity Score. No deaths, malignancies, opportunistic infections, or tuberculosis cases occurred. Safety events were consistent with the known ustekinumab safety profile.
Conclusion Of the 46 patients who entered the voluntary extension of this phase II study, clinical benefit in global and organ-specific SLE activity measures was observed with ustekinumab through 2 years with no new or unexpected safety findings. [ClinicalTrials.gov: NCT02349061]
The disease burden of systemic lupus erythematosus (SLE) can be substantial, with symptoms affecting the skin, hair, joints, kidneys, and central nervous system.1 It has been estimated that nearly 80% of patients will experience skin lesions,2 arthritis,3 or cognitive impairment,4 and approximately half will develop renal disease.5 Other complications of SLE include alopecia, vasculitis, fibromyalgia, seizures, myelitis, and neuropathy.1 Improvements in treatments over time have resulted in longer survival times; however, patients with SLE continue to have a higher mortality rate than the general population.6 Long-term treatment response to conventional therapies, including corticosteroids and immunosuppressants, remains suboptimal owing to incomplete disease control and treatment toxicities. Treatment options for patients with SLE remain limited, with only 3 new treatments approved in recent decades.7,8,9 Thus, there remains a high unmet need for effective therapies for SLE to reduce morbidity and mortality with improved tolerability.
Ustekinumab is a fully human monoclonal antibody that inhibits the p40 subunit of interleukin (IL)-12 and IL-23 and is approved for the treatment of plaque psoriasis, psoriatic arthritis, Crohn disease, and ulcerative colitis. In a phase II study evaluating the safety and efficacy of ustekinumab in patients with SLE, greater improvement in disease activity measures were observed with ustekinumab compared with placebo at week 24,10 and improvements were sustained through the first year.11 Here, we report the final efficacy and safety results through 2 years from the open-label extension of this randomized trial.
METHODS
Patients and study design. Details of the patient eligibility criteria and study design for this phase II, randomized, placebo-controlled study have been previously described.10,11 Briefly, adults aged 18–75 years who met Systemic Lupus International Collaborating Clinics classification criteria for SLE12 for ≥ 3 months before the first study drug administration were eligible. Patients had to have a Systemic Lupus Erythematosus Disease Activity Index 2000 (SLEDAI-2K) score13 ≥ 6 at screening, and a SLEDAI-2K score ≥ 4 for clinical features (excluding laboratory results) at baseline. Eligible patients also had ≥ 1 British Isles Lupus Assessment Group 2004 index (BILAG) A (severe manifestation) or 2 BILAG B (moderate manifestation) organ domain scores14 during screening. In addition, patients were required to have at least 1 positive test for autoantibodies (antinuclear antibody titer ≥ 1:80 by immunofluorescence, and/or anti-dsDNA antibodies > 75 IU/mL, and/or anti-Smith antibodies > 120 AU/mL) at screening, as well as a documented medical history of a previous positive titer for ≥ 1 of these autoantibodies.
Patients were randomly assigned (3:2) to receive a single intravenous (IV) infusion of ustekinumab (260 mg for patients weighing ≥ 35 kg to ≤ 55 kg; 390 mg for patients weighing > 55 kg to ≤ 85 kg; 520 mg for patients weighing > 85 kg) at week 0, followed by SC injections of ustekinumab 90 mg at week 8 and every 8 weeks thereafter through week 40, or placebo infusion at week 0 followed by placebo SC injections at week 8 and every 8 weeks thereafter. Patients in the placebo group crossed over to ustekinumab at week 24 and began SC injections of ustekinumab 90 mg every 8 weeks from week 24 through week 40 without receiving an initial IV dose. After week 40, patients could enter the optional open-label extension. The study extension was not planned as part of the initial study design; thus, some patients did not enter the study extension as they had already completed their study participation. Eligible patients participating in the study extension could enter either at week 48 or week 56 and receive SC ustekinumab every 8 weeks through week 104.
All patients were required to be receiving ≥ 1 background therapy for SLE prior to screening. Stable doses of the following concomitant medications were permitted during the study: oral glucocorticoids (GCs; ≤ 20 mg/day, prednisone or equivalent) for ≥ 4 weeks before first study agent administration; immunosuppressant drugs (eg, mycophenolate mofetil/mycophenolic acid ≤ 2 g/day, azathioprine/6-mercaptopurine ≤ 2 mg/kg/day, or methotrexate ≤ 25 mg/wk) for ≥ 6 weeks; antimalarials (eg, hydroxychloroquine, quinacrine, or chloroquine) for ≥ 8 weeks with a stable dose for ≥ 6 weeks; or angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, nonsteroidal antiinflammatory drugs, other analgesics, or topical low or medium potency GCs for ≥ 2 weeks.
Assessments. Global clinical efficacy assessments included the SLEDAI-2K Responder Index-4 (SRI-4) response, which is a composite measure requiring ≥ 4-point reduction from baseline in SLEDAI-2K score, no worsening (< 10% increase) in the physician global assessment (PGA)15 score from baseline, and no new BILAG A and no more than 1 new BILAG B domain scores.14 SRI-5 and SRI-6 responses were assessed similarly to the SRI-4 response, with the exception that SLEDAI-2K improvement from baseline is ≥ 5 or 6 points, respectively. SLEDAI-2K response (≥ 4-point improvement from baseline), PGA response (≥ 30% improvement from baseline), and BILAG-based Combined Lupus Assessment (BICLA) response16 were also used to assess overall efficacy. BICLA response is a composite measure requiring BILAG improvement (all BILAG A and B scores present at baseline improve to lower grade scores, no new BILAG A domain scores, and ≤ 1 new BILAG B domain score), no worsening in PGA (< 10% increase), no worsening in SLEDAI-2K score from baseline (change ≤ 0), and meeting no treatment failure criteria.
Mucocutaneous disease was evaluated using the Cutaneous Lupus Erythematosus Disease Area and Severity Index (CLASI),17 with CLASI response defined as ≥ 50% improvement from baseline CLASI Activity Score in patients with a baseline CLASI Activity Score ≥ 4. Joint disease was evaluated by recording the numbers of tender, swollen, and active joints (defined as having both pain/tenderness and signs of inflammation, such as swelling or effusion). Joint response was defined as ≥ 50% improvement from baseline active joint count in patients with ≥ 4 active joints at baseline. Flare was assessed using the BILAG-2004 criteria18 and was defined as ≥ 1 new BILAG A domain score (severe flare) or ≥ 2 new BILAG B domain scores (moderate flare) compared with baseline. Health-related quality of life (HRQOL) was assessed using the 36-item Short Form Health Survey (SF-36); minimum clinically important difference [MCID] was defined as improvements of ≥ 5 points in individual domains and ≥ 2.5 points in the physical component summary (PCS) and mental component summary (MCS) scores.19,20
Safety was monitored throughout the study, and adverse events (AEs) were reported according to the actual treatment received. The final safety assessment was at week 120. Serum samples were collected at weeks 0, 4, 8, 12, 16, 24, 40, 48, 80, 104, 112, and 120 for evaluating the presence of antibodies to ustekinumab; samples were also collected at 8 and 16 weeks after the last study agent administration for patients who discontinued study treatment before week 40. Immunogenicity was assessed using a highly sensitive, drug-tolerant, enzyme immunoassay.
This trial was conducted in accordance with the principles of the Declaration of Helsinki and Good Clinical Practices (ClinicalTrials.gov: NCT02349061). The protocol was approved by a central institutional review board (IRB; Sterling in the US; approval number: 5002C) or local IRBs or Ethics Committees at individual sites. Patients gave written informed consent before any study-related procedures were performed.
Statistical analysis. Efficacy results are reported according to randomized treatment group through week 112. For patients randomized to receive ustekinumab, efficacy analyses included all patients who were randomized to receive ustekinumab at baseline and received ≥ 1 ustekinumab administration. For patients randomized to receive placebo, efficacy analyses after the placebo-controlled period included only patients who crossed over to ustekinumab at week 24. No formal hypothesis testing was performed between the treatment groups after week 24. BICLA response rates were determined using the intent-to-treat population, and patients who met the treatment failure criteria10 before week 24 were classified as nonresponders for subsequent timepoints through week 112. Treatment failure rules were applied through week 48 for the proportion of patients achieving SRI and SLEDAI responses, with observed data reported during the extension. Through week 48, patients who withdrew from the study or had missing values were considered to be nonresponders. For the proportion of patients achieving PGA, CLASI, and joint response and the proportion of patients with no worsening in BILAG or PGA scores, values for patients who met the treatment failure criteria were classified as missing data from the point of treatment failure forward through week 48, with observed data reported for the study extension. Mean changes from baseline in PGA and SLEDAI-2K scores and SF-36 PCS and MCS outcomes were analyzed using observed data.
RESULTS
Patient disposition. At baseline, 102 patients were randomly assigned to receive placebo (n = 42) or ustekinumab (n = 60). Baseline demographic and disease characteristics were well balanced between the treatment groups.10 Through week 24, 9 patients in the placebo group and 4 in the ustekinumab group discontinued study treatment. Thirty-three patients from the placebo group crossed over to receive ustekinumab at week 24.10 A total of 83 patients (placebo crossover, n = 30; ustekinumab, n = 53) completed treatment through week 40, which was the final ustekinumab administration of the main study.11 Forty-six patients entered the optional study extension (placebo crossover, n = 17; ustekinumab, n = 29), 26 patients entered the extension at week 48, and 20 patients entered at week 56 (Figure 1). Of these, 8 patients discontinued treatment before week 104, with the most common reasons being AEs (n = 3) and withdrawal of consent (n = 3).
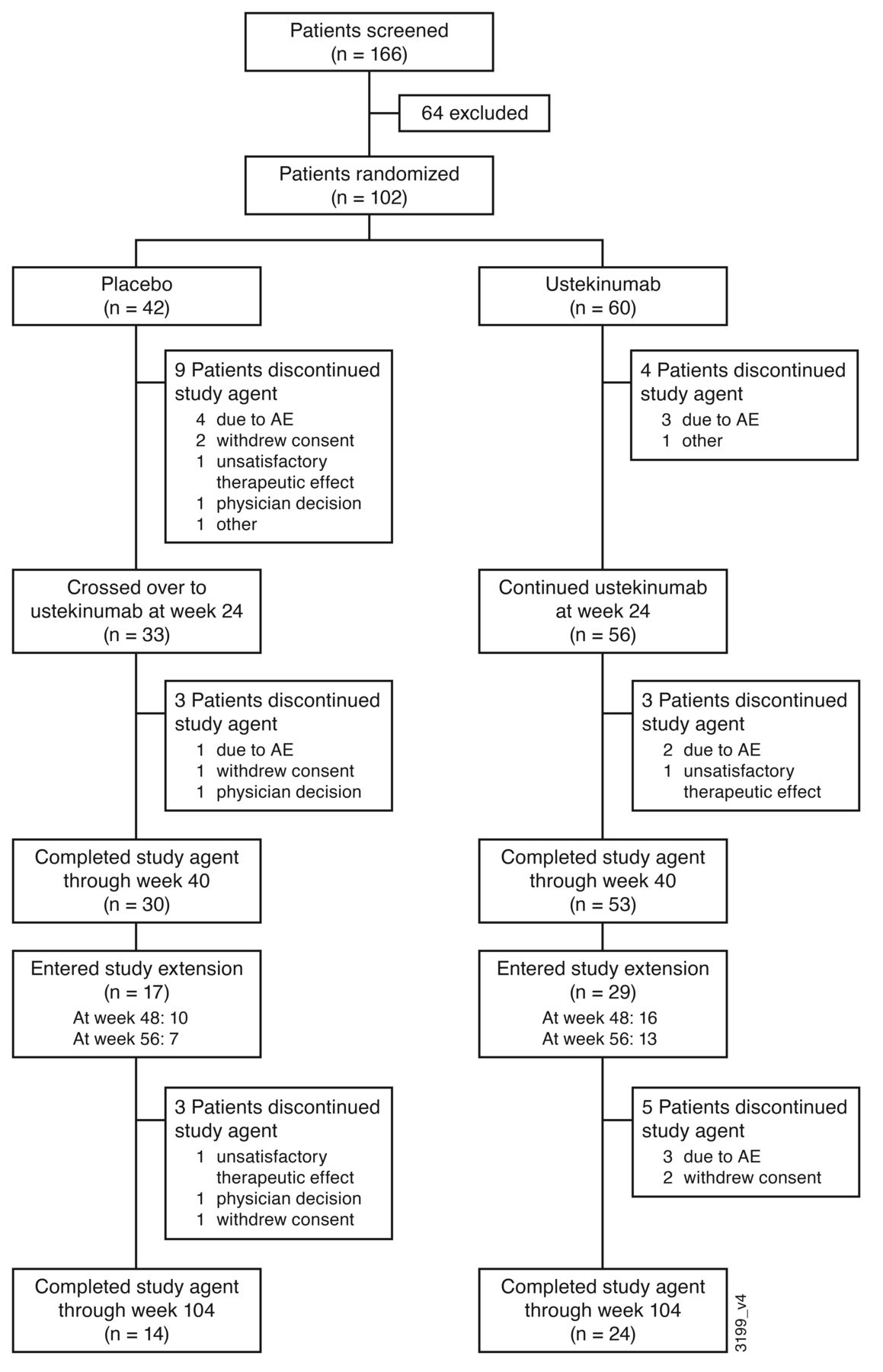
Patient disposition through week 120. AE: adverse event.
Efficacy. The primary endpoint of the study, at week 24, was achieved: 62% of patients in the ustekinumab group had an SRI-4 response without meeting treatment failure criteria compared with 33% of patients in the placebo group.10 At 1 year, the proportion of patients achieving SRI-4 response was maintained among patients in the ustekinumab group (63%); of the 33 patients who crossed over from placebo to ustekinumab at week 24, 42% had an SRI-4 response.11 Among the 46 patients who entered the study extension, 79% (19 of 24) in the ustekinumab group and 92% (12 of 13) in the placebo crossover group had an SRI-4 response at week 112 (Figure 2). Similar trends were observed for SRI-5 and SRI-6 response (Figure 2). In general, improvements in SLEDAI-2K score, joint, and mucocutaneous disease measures were also greater in the ustekinumab group compared with placebo at week 2410 and maintained through 1 year.11 At week 112, 92% in both the ustekinumab (22 of 24) and the placebo crossover (12 of 13) groups had ≥ 4-point improvement from baseline in SLEDAI-2K score; 79% (19 of 24) and 93% (13 of 14), respectively, had ≥ 30% improvement from baseline in PGA; 86% (12 of 14) and 91% (10 of 11), respectively, had ≥ 50% improvement in active joint (pain and inflammation) count; and 79% (11 of 14) and 100% (5 of 5), respectively, had ≥ 50% improvement in CLASI Activity Score (Figure 3). The proportion of patients who had a BICLA response was maintained through the study extension (Supplementary Figure 1, available with the online version of this article).
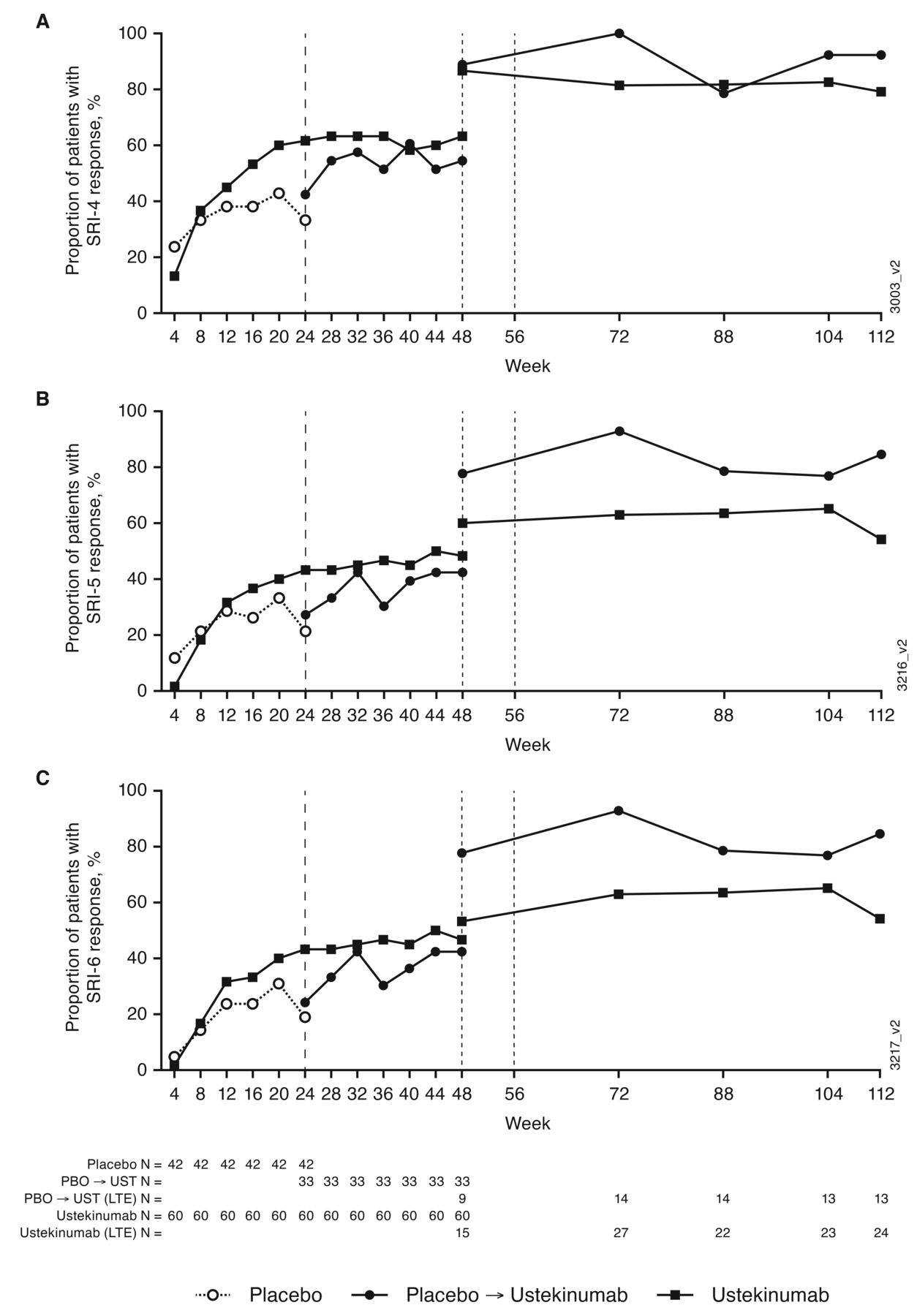
Proportions of patients with (A) SRI-4, (B) SRI-5, and (C) SRI-6 responses through week 112. Patients participating in the study extension could have entered either at week 48 or week 56. SRI was defined as ≥ 4-/5-/6-point reduction in SLEDAI-2K total score, no new BILAG A and no more than 1 new BILAG B domain scores and no worsening (< 10% increase) from baseline in the physician global assessment. BILAG: British Isles Lupus Assessment Group 2004 index; LTE: long-term extension; PBO: placebo; SLE: systemic lupus erythematosus; SLEDAI-2K: Systemic Lupus Erythematosus Disease Activity Index 2000; SRI-4/5/6: SLE responder index; UST: ustekinumab.
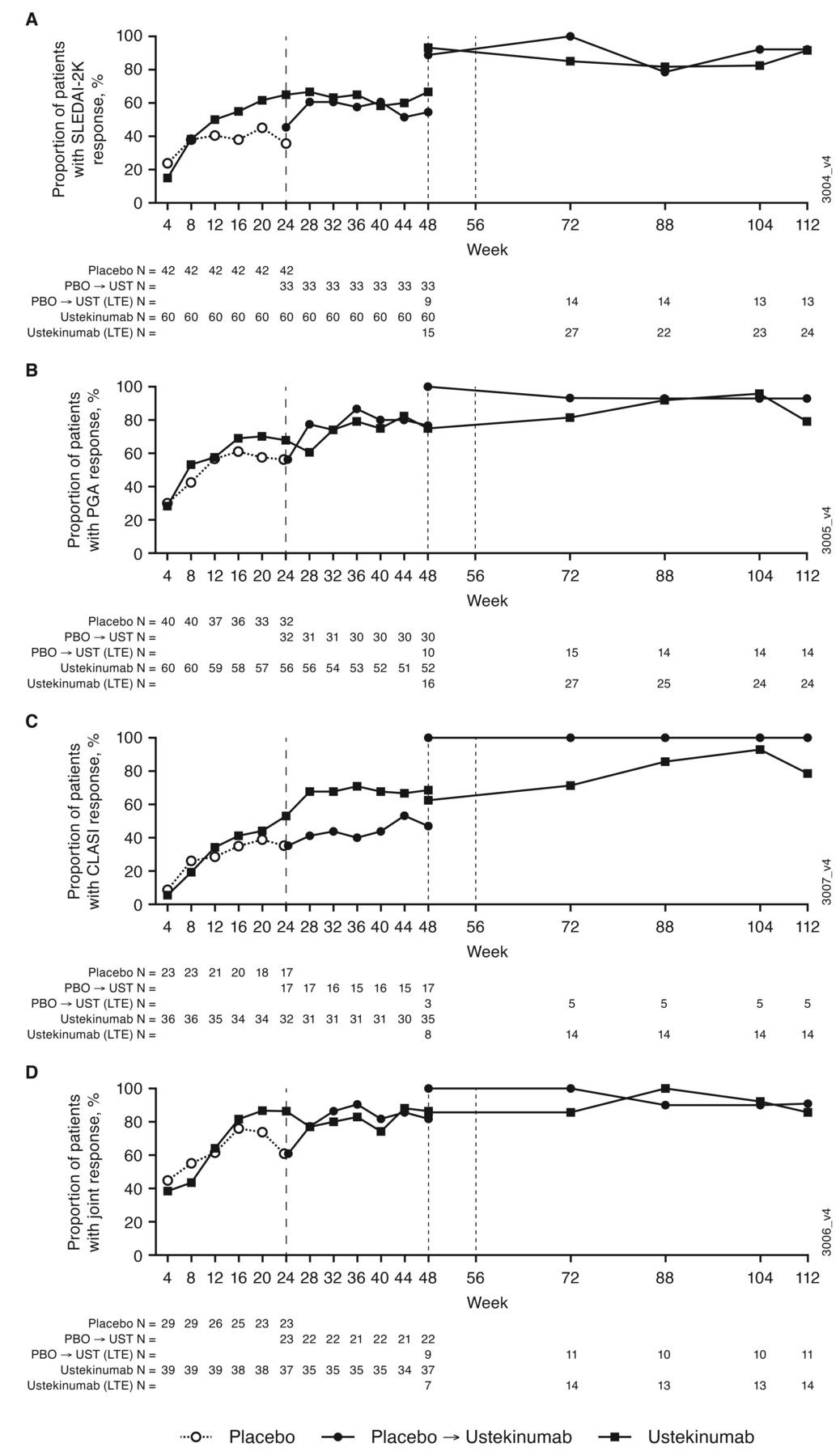
Proportions of patients with (A) SLEDAI-2K, (B) PGA, (C) CLASI Activity Score, and (D) joint responses. Patients participating in the study extension could have entered either at week 48 or week 56. SLEDAI-2K response defined as ≥ 4-point improvement from baseline; PGA response defined as ≥ 30% improvement from baseline; CLASI response defined as ≥ 50% improvement from baseline in patients with a baseline CLASI Activity Score ≥ 4; joint response defined as ≥ 50% improvement from baseline in the number of joints with pain and inflammation (active joints) in patients with ≥ 4 active joints at baseline. CLASI: Cutaneous Lupus Erythematosus Disease Area and Severity Index; LTE: long-term extension; PBO: placebo; PGA: physician global assessment; SLEDAI-2K: Systemic Lupus Erythematosus Disease Activity Index 2000; UST: ustekinumab.
During the study extension, 2 patients experienced a flare; 1 patient had a new BILAG A flare (at week 88 and during the 8-week follow-up period) in the mucocutaneous domain, and 1 patient had ≥ 2 BILAG B flares (at week 64) in the constitutional and mucocutaneous domains. The incidence per 10,000 patient-days of a BILAG 1A/2B flare was 0 in the placebo crossover group and 1.95 in the ustekinumab group.
Mean improvements in all domains of the SF-36 except vitality were observed in the ustekinumab treatment group from weeks 8–24, and in all SF-36 domains from weeks 16–24 in the placebo treatment group. At week 24, numerically larger mean improvements were observed in the ustekinumab treatment group for 5 of the 8 SF-36 domains (bodily pain, mental health, physical functioning, role emotional, and social functioning) than the placebo treatment group. Improvements ≥ 2.5 points (the MCID for PCS and MCS summary scores19, 20) in SF-36 PCS scores were observed and maintained from weeks 48–72 in the ustekinumab group and from weeks 48–104 in the placebo crossover group. Mean improvements in SF-36 MCS scores ≥ 2.5 points were observed at week 24 and week 72 in the ustekinumab group, and at week 24 and from weeks 48 to 104 in the placebo crossover group (data not shown).
Safety. Through week 120 (final safety assessment), 93 patients received ≥ 1 administration of ustekinumab; 60 who were randomized to receive ustekinumab at baseline and 33 in the placebo group who crossed over to ustekinumab at week 24 (Table 1). Through week 120, 80 (86%) patients treated with ustekinumab experienced ≥ 1 AE, with infections being the most common type of AE. Among patients treated with ustekinumab, the most common AEs were upper respiratory tract infection (n = 23, 22.5%), urinary tract infection (n = 21, 20.6%), and nasopharyngitis (n = 14, 13.7%).
Treatment-emergent AEs through week 120.
Through week 120, serious AEs (SAEs) occurred in 17 (18.3%) patients treated with ustekinumab. Nine patients treated with ustekinumab experienced a total of 11 serious infections; 9 serious infections occurred prior to week 56 and have been reported previously.10,11 Two patients had a serious infection during the study extension period: a 66-year-old patient was hospitalized with a viral infection and recovered, and a 21-year-old patient was diagnosed with sinusitis and later recovered. Two major cardiovascular AEs occurred in patients treated with ustekinumab. Prior to week 24, 1 patient had an ischemic stroke; this patient was aged 56 years and had multiple risk factors at baseline including hypertension, tobacco use, and long-term GC use.10 A second patient had right coronary artery occlusion during the study extension; this patient was aged 66 years with several risk factors at baseline, including history of angioplasty, atrial fibrillation, blocked circumflex artery, hypertension, hyperlipidemia, and ischemic heart disease. Both events were assessed by the investigators as not related to ustekinumab. Other SAEs that occurred during the study extension were exacerbation of Raynaud phenomenon (n = 1) and worsening of SLE and lupus nephritis (both in the same patient). No cases of active tuberculosis, opportunistic infections, malignancies, or deaths were reported in the study.
Infusion reactions were only reported through week 24 as infusions were administered only at baseline. One patient in the ustekinumab group experienced an infusion reaction (suspected anaphylactic reaction) and discontinued study treatment.10 Another patient in the ustekinumab group experienced multiple mild injection site reactions during the main study10; no injection site reactions occurred during the study extension.
Immunogenicity. Ninety-two patients (placebo crossover, n = 32; ustekinumab, n = 60) received ≥ 1 administration of ustekinumab and had ≥ 1 serum sample available after their first ustekinumab administration and were included in the immunogenicity analyses. Through week 120, a total of 18 (19.6%) patients treated with ustekinumab tested positive for antibodies to ustekinumab (placebo crossover, n = 11 of 32 [34.4%]; ustekinumab, n = 7 of 60 [11.7%]). Of these, 5 patients (placebo crossover, n = 3; ustekinumab, n = 2) had antibody titers > 1:1000. Seven patients treated with ustekinumab tested positive for neutralizing antibodies; no effect on clinical response was observed and none of these patients had an allergic reaction.
DISCUSSION
Previous results from this phase II, placebo-controlled trial showed that a significantly greater proportion of patients treated with ustekinumab had an SRI-4 response without meeting treatment failure criteria at week 24 compared with placebo.10 Additionally, placebo patients who crossed over to ustekinumab at week 24 demonstrated improvements in global disease activity as measured by SLEDAI-2K, PGA, number of active joints, and CLASI Activity Score at 1 year.11 Among the patients who entered the optional study extension, 12 of 13 patients in the placebo crossover group and 19 of 24 in the ustekinumab group had an SRI-4 response at week 112. Two patients experienced a flare during the extension period. Improvements in HRQOL PCS score were numerically greater in the ustekinumab group than in the placebo group through the placebo-controlled period. Beyond the placebo-controlled period, clinically meaningful improvements were observed at 1 year11 and through the end of the study extension at 2 years.
Through week 120, 86% of all patients treated with ustekinumab had at least 1 AE. Among these, infections were the most common. A total of 17 patients treated with ustekinumab had an SAE. Two major cardiovascular AEs occurred during the study, both in patients with multiple risk factors, including hypertension, hyperlipidemia, tobacco use, atrial fibrillation, and ischemic heart disease. Through 2 years, there were no cases of active tuberculosis, opportunistic infections, malignancies, or deaths. The safety results through week 120 were consistent with the known safety profile of ustekinumab, and no new or unexpected safety signals were identified.21
Through week 120, the proportion of patients with antibodies to ustekinumab was numerically higher in the patients who crossed over from placebo to ustekinumab (34.4%) than in patients who had been receiving ustekinumab from baseline (11.7%), which was consistent with results reported through week 56 (10 of 32 [31.3%] and 6 of 60 [10.0%], respectively).11 Of note, patients in the placebo crossover group only received the SC injections of ustekinumab, unlike the patients randomized to ustekinumab who received the induction IV infusion followed by maintenance SC injections. The immunogenicity findings from this study are consistent with the effect of high-zone tolerance that has been previously demonstrated.22,23 In addition, different routes of administration for an initial drug exposure (ie, IV infusion or SC injection) can elicit distinct immune responses that may affect development of antidrug antibodies.24
Results of this study through 2 years are limited by the small number of patients who entered the study extension. Some patients had completed their study participation when the extension was added to the study protocol; in total, 46 patients opted to enter the open-label extension. The voluntary nature of the study extension may have introduced attrition bias in the results reported past 1 year. In addition, results could have also been influenced by responder bias, with those patients who demonstrated improvements in disease activity being more likely to remain in the study. It should be noted that the phase III study (ClinicalTrials.gov: NCT03517722) of ustekinumab in patients with SLE was discontinued early following a preplanned interim analysis because of a lack of efficacy, and there were no new safety signals leading to the decision. Additional analyses will be performed once the totality of the phase III study data is available to evaluate potential reasons for the different results of the 2 studies.
In summary, clinical benefit in global and organ-specific SLE activity measures was observed for patients receiving ustekinumab through 2 years in this voluntary, open-label extension of a phase II study, with no new or unexpected safety findings.
ACKNOWLEDGMENT
The authors thank Rebecca Clemente, PhD, of Janssen Scientific Affairs, LLC, for writing support.
Footnotes
This study was funded by Janssen Research & Development, LLC.
M. Chevrier passed away on February 6, 2021.
RFvV has received consulting fees, speaking fees, and/or honoraria from AbbVie, AstraZeneca, Biotest, BMS, Celgene, Eli Lilly, GSK, Janssen, Medac, Merck, Novartis, Pfizer, Roche, and UCB, and research support from AbbVie, Arthrogen, BMS, Eli Lilly, GSK, Pfizer, and UCB. BHH has received consulting fees, speaking fees, and/or honoraria from Aurinia, GSK, and UCB. GCT has received consulting fees from A2 Therapeutics and research support from Janssen. PL has received consulting fees from Janssen. RMG, KF, KHL, MC, SR, ZY, CSK, and QZ are or were employees of Janssen Research & Development, LLC, when this work was performed and own stock in Johnson & Johnson, of which Janssen Research & Development, LLC, is a wholly owned subsidiary. PB is an employee of Janssen Pharmaceutical Companies of Johnson & Johnson and owns stock in Johnson & Johnson.
- Accepted for publication November 19, 2021.
- Copyright © 2022 by the Journal of Rheumatology
This is an Open Access article, which permits use, distribution, and reproduction, without modification, provided the original article is correctly cited and is not used for commercial purposes.








{kind=link}
{kind=link}
{kind=link}