

Abstract
Objective To assess the efficacy of secukinumab on axial and peripheral enthesitis in patients with ankylosing spondylitis (AS) using pooled data from randomized controlled phase III studies.
Methods In this posthoc analysis, data were pooled from patients originally randomized to secukinumab 150 mg, 300 mg, or placebo (PBO) from phase III MEASURE 1–4 studies (ClinicalTrials.gov: NCT01358175, NCT01649375, NCT02008916, and NCT02159053). Maastricht AS Enthesitis Score (MASES) was used for assessments of enthesitis through Week 52. Efficacy outcomes were mean change in MASES score and complete resolution (MASES = 0) of enthesitis in patients with baseline MASES > 0.
Results A total of 693 (71.5%) patients had enthesitis at baseline in secukinumab 300 mg, 150 mg, and PBO groups (58 [76.3%], 355 [70.4%], and 280 [72%], respectively) out of 969 patients pooled in this analysis. At Week 16, mean changes from baseline for overall MASES and enthesitis at axial MASES sites, respectively, were as follows: –2.9 (P < 0.01) and –2.9 (P < 0.01) for secukinumab 300 mg; –2.4 (P < 0.015) and –2.3 (P < 0.05) for secukinumab 150 mg; and –1.9 and –1.8 for PBO, with improvements seen through Week 52. More than one-third of secukinumab-treated patients (300 mg: 36.2%; 150 mg: 40.8%) achieved complete resolution of enthesitis at Week 16.
Conclusion Secukinumab improved enthesitis at overall MASES and axial sites in patients with AS.
Enthesitis, which is the inflammation of the entheses, is a hallmark feature of ankylosing spondylitis (AS; also termed, radiographic axial spondyloarthritis [axSpA]), a chronic and inflammatory form of arthritis. It is a distinctive pathological manifestation characterized by inflammation at the tendon, ligament, or joint capsule insertion sites to bones.1 Enthesitis is often associated with considerable pain, morning stiffness, fatigue, and impaired physical function, frequently leading to poor quality of life.2,3
The pathogenesis of enthesitis is not fully understood. Enthesitis is triggered by biomechanical stress or infection, which induces the activation of γδ T cells and type 3 innate lymphoid cells at entheseal sites. This subsequently stimulates the production of tumor necrosis factor (TNF), interleukin (IL)17A, and IL-23, leading to the influx and activation of neutrophils, which in turn leads to entheseal inflammation.3,4 This IL-driven enthesitis may cause irreversible structural damage in patients with AS.5,6
Enthesitis can encompass peripheral and/or axial manifestations. Peripheral enthesitis is frequently observed in the lower limbs (e.g., muscle attachments to the greater and lesser trochanters, the insertion of the quadriceps tendon at the upper patellar pole, the patellar tendon insertion on the tibial plateau, and the Achilles tendon insertion into the calcaneus), whereas axial enthesitis sites mainly include the thoracic and lumbar spine and costosternal joints.7,8,9 Different indices have been developed for the clinical evaluation of enthesitis in AS, including the Mander Enthesitis Index, the Modified Mander Enthesitis Index,10 the Major Enthesitis Index,11 the Maastricht AS Enthesitis Score (MASES),12 and the SpA Research Consortium of Canada (SPARCC) enthesitis score.13
According to the Corrona registry, patients with axSpA who suffer from enthesitis have a higher disease burden than those without enthesitis.14 Although the prevalence of enthesitis among patients with AS ranges from 30–70%, enthesitis is often under diagnosed in routine practice.15,16,17 In clinical practice, the aim of enthesitis treatment is to resolve entheseal-related pain and inflammation.3 Nonsteroidal antiinflammatory drugs (NSAIDs) are the recommended first-line treatment for patients with AS and have been demonstrated to be effective in reducing inflammation and pain in peripheral enthesitis.18,19,20,21 However, in patients with long-standing enthesitis, as opposed to those with a more recent onset, NSAIDs may not adequately control the disease.3 In patients with axSpA who do not respond adequately or are intolerant to NSAIDs, treatment with TNF inhibitors (TNFi) is indicated and has been shown to be effective in the treatment of enthesitis. Although TNFi significantly improves the signs and symptoms in patients with AS,22,23,24,25,26 approximately 40–50% of patients experience either inadequate response, intolerance, or safety concerns with TNFi therapy.26–34
Secukinumab, a human monoclonal IgG1κ antibody that directly inhibits IL-17A, has been demonstrated to provide rapid and sustained improvement in the signs and symptoms of active AS.35,36,37,38,39 It has also been shown to provide rapid and sustained resolution of enthesitis in approximately 50–70% of patients with psoriatic arthritis (PsA).40,41,42 However, data on the effect of secukinumab on the improvement or resolution of enthesitis in patients with AS are limited. Herein, we present a posthoc analysis using data pooled from 4 phase III studies (MEASURE 1–4) over 52 weeks that had data available at the reporting cutoff date of May 13, 2018. The aim of this posthoc analysis was to determine the efficacy of secukinumab vs placebo (PBO) on mean change from baseline in MASES score and complete resolution of enthesitis (MASES = 0) in patients with AS with enthesitis at baseline.
METHODS
Study design and patients. MEASURE 1 (NCT01358175) and MEASURE 4 (NCT02159053) were 2-year studies, whereas MEASURE 2 (NCT01649375) and MEASURE 3 (NCT02008916) were 5- and 3-year randomized, double-blind, placebo-controlled phase III studies. The details of the study designs, methods, and results have been reported previously.35,36,37,38 Detailed inclusion and exclusion criteria are listed in Supplementary Table 1 (available with the online version of this article). Briefly, in MEASURE 1 and MEASURE 3, patients were randomized to intravenous (IV) secukinumab 10 mg/kg or PBO at baseline, Week 2, and Week 4, followed by subcutaneous (SC) secukinumab 150 mg or 75 mg in MEASURE 1 or SC secukinumab 300 mg or 150 mg in MEASURE 3 vs matched PBO every 4 weeks (Q4W), starting at Week 8. In MEASURE 2 and MEASURE 4, patients were randomized to SC secukinumab 150 mg or 75 mg (MEASURE 2) or SC secukinumab 150 mg with or without loading doses (MEASURE 4) vs matched PBO at baseline, Weeks 1, 2, and 3, and Q4W starting at Week 4. In MEASURE 2–4, PBO-treated patients were rerandomized at Week 16 to SC secukinumab Q4W, regardless of clinical response, whereas in MEASURE 1, rerandomization from PBO to secukinumab occurred at Week 16 in nonresponders failing to achieve Assessment of Spondyloarthritis international Society 20 (ASAS20) criteria and at Week 24 in responders.35,36,37,38 These multicenter randomized clinical trials (RCTs) were well designed and fulfilled the Consolidated Standards of Reporting Trials Statement checklist, which comprises a minimum standard of recommendations for reporting RCTs.43
The pooled patient populations from the MEASURE 1–4 studies were considered in this posthoc exploratory analysis. Data were pooled from patients originally randomized to secukinumab 150 mg, 300 mg (MEASURE 3 only), or PBO, with enthesitis at baseline defined as MASES > 0. Patients on the same dose of secukinumab were pooled across studies, regardless of loading regimen (IV or SC). Analysis included all patients with baseline entheseal tenderness and/or swelling of at least 1 out of the 13 MASES sites (the bilateral first and seventh costochondral joints, the anterior and posterior superior iliac spines, the iliac crests, the proximal insertions of Achilles tendon, and the fifth lumbar spinous process [overall score range 0–13]), as well as subgroup analyses of the 11 axial entheseal sites (13 MASES sites minus 2 Achilles tendons), 6 peripheral entheseal sites (2 Achilles tendons + 4 lateral condyles of both humeri/femurs), and the 2 Achilles tendons separately (Figure 1).
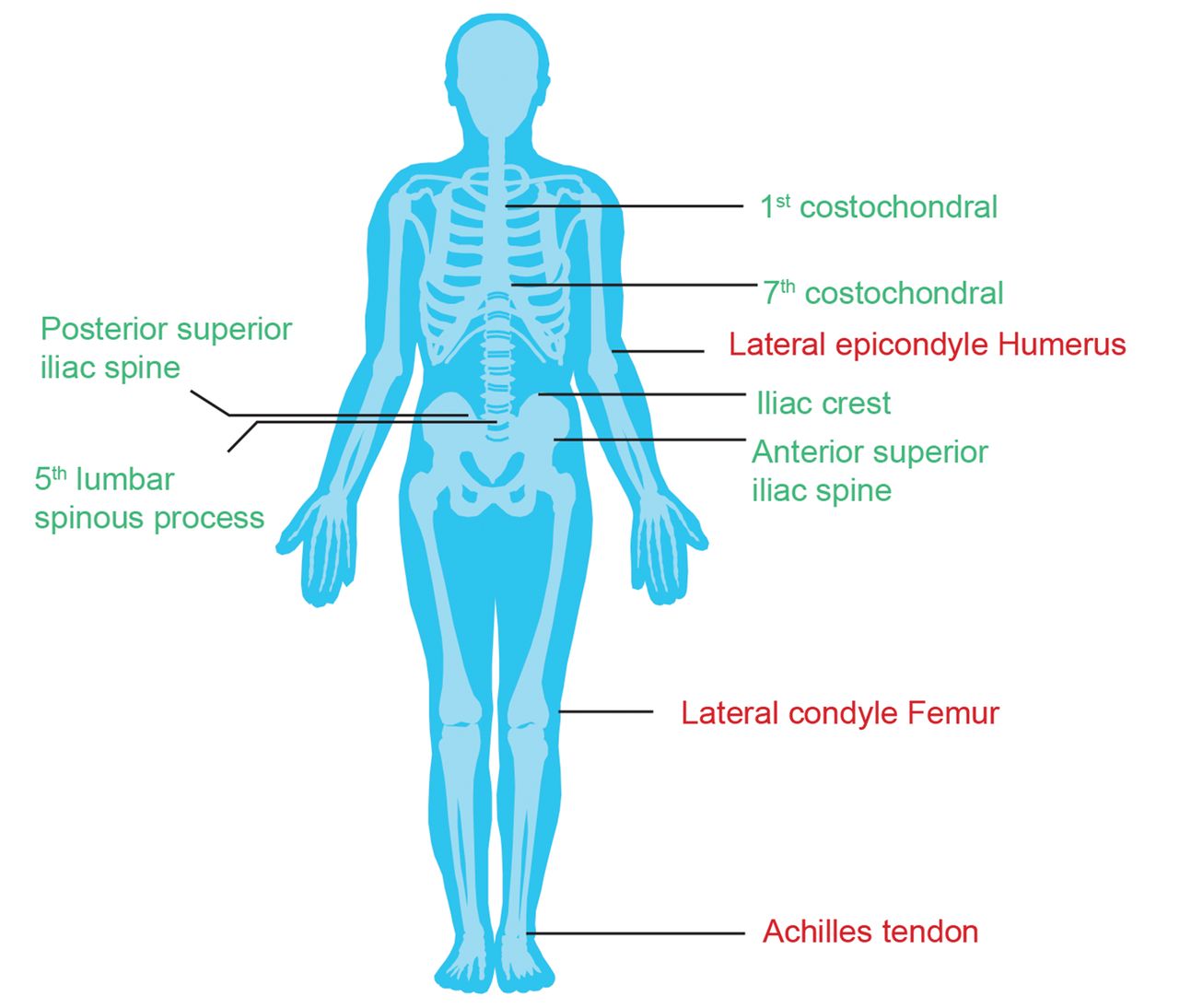
Major entheseal sites. Overall MASES entheseal sites (13 sites), defined as: first costochondral (R, L) + seventh costochondral (R, L) + posterior superior iliac spine (R, L) + anterior superior iliac spine (R, L) + iliac crest (R, L) + proximal Achilles tendon (R, L) + fifth lumbar spinous process. Achilles Tendon (2 sites), defined as: proximal Achilles tendon (R, L). Axial entheseal sites (in green; 11 sites), defined as: first costochondral (R,L) + seventh costochondral (R,L) + posterior superior iliac spine (R, L) + anterior superior iliac spine (R,L) + iliac crest (R,L) + fifth lumbar spinous process. Peripheral entheseal sites (in red; 6 sites), defined as: proximal Achilles tendon (R, L) + lateral epicondyle humerus (R, L) + lateral condyle femur (R, L). L: left; MASES: Maastricht Ankylosing Spondylitis Enthesitis Score; R: right.
All the studies were conducted in compliance with the ethical principles originating in or derived from the Declaration of Helsinki and in compliance with all International Conference on Harmonization Good Clinical Practice guidelines. All patients signed an informed consent form that was reviewed and approved by an independent ethics committee or institutional review board.35,36,37,38 Ethics approval numbers for the main institutions are included in Supplementary Table 2 (available with the online version of this article).
In all the 4 studies, patients aged ≥18 years with AS fulfilling the modified New York criteria44 and active disease as defined by Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) ≥ 4 (range 0–10)45 and a spinal pain score of ≥ 4 cm on a visual analog scale (0–10 cm), despite treatment with maximal tolerated doses of NSAIDs were included. Patients had either an inadequate response or were intolerant to no more than 1 TNFi agent.36,46 Patients previously treated with no more than 1 TNFi were included if they had an inadequate response to an approved dosage for ≥ 3 months or were intolerant to at least 1 dose (hereafter collectively referred to as patients with an inadequate response to TNFi [TNFi-IR]). Patients could continue to receive the following medications at a stable dose: sulfasalazine (≤ 3 g/d), methotrexate (7.5–25 mg/week), prednisone or equivalent (≤ 10 mg/d), and NSAIDs.
Key exclusion criteria were total spinal ankylosis, evidence of infection or cancer on chest radiography, active systemic infection within 2 weeks before the baseline visit, and previous treatment with cell-depleting therapies or biologics other than TNFi agents.
Efficacy outcomes. In this posthoc analysis, the efficacy endpoints assessed at Week 52 were the mean change from baseline in MASES score and complete resolution (MASES = 0) in patients with enthesitis at baseline.
Statistical analysis. A mixed-effect model for repeated measures (MMRM) analysis was done for change from baseline in MASES score for patients with enthesitis at baseline. Only patients with MASES > 0 at baseline were included in the analysis to avoid misrepresentation of a potential treatment effect from the approximately 30% of patients with no enthesitis at baseline who would, therefore, be expected to have low enthesitis scores during treatment as well. MMRM included treatment, TNFi-IR status (yes/no), and scheduled visits as fixed effects, with baseline score and weight as covariates. Treatment and baseline by visit were included as interaction terms in the model. The effects of secukinumab 300 mg and 150 mg were analyzed using a logistic regression model for resolution of enthesitis through Week 16. Logistic regression used treatment as a factor, with baseline score and weight as covariates. These analyses were also performed for different entheseal sites (namely, overall MASES entheseal sites, axial entheseal sites, the Achilles tendons, and other peripheral entheseal sites). Missing values, including those due to discontinuation of study treatment, were imputed as nonresponse through Week 16. Data are reported as observed thereafter up to Week 52.
RESULTS
Baseline characteristics and patient demographics. A total of 969 patients were included in this analysis, of which 693 (71.5%) patients had enthesitis at baseline (secukinumab 300 mg: 76.3% [58/76]; secukinumab 150 mg: 70.4% [355/504]; and PBO: 72.0% [280/389]; Figure 2). Baseline demographics were generally comparable across groups except for a higher proportion of females among the patients with enthesitis (39.7%, 39.2%, and 39.6% in secukinumab 300 mg, 150 mg, and PBO arms, respectively) vs patients without enthesitis (16.7%, 16.8%, and 19.3% in secukinumab 300 mg, 150 mg, and PBO arms, respectively). In line with the objective of this analysis, we only report data for patients with enthesitis at baseline (Table 1). The baseline characteristics of patients without enthesitis have been presented in Supplementary Table 3 (available with the online version of this article).

Patient population of the pooled analysis from MEASURE 1–4 studies.
Demographics and baseline characteristics of patients with enthesitis at baseline.
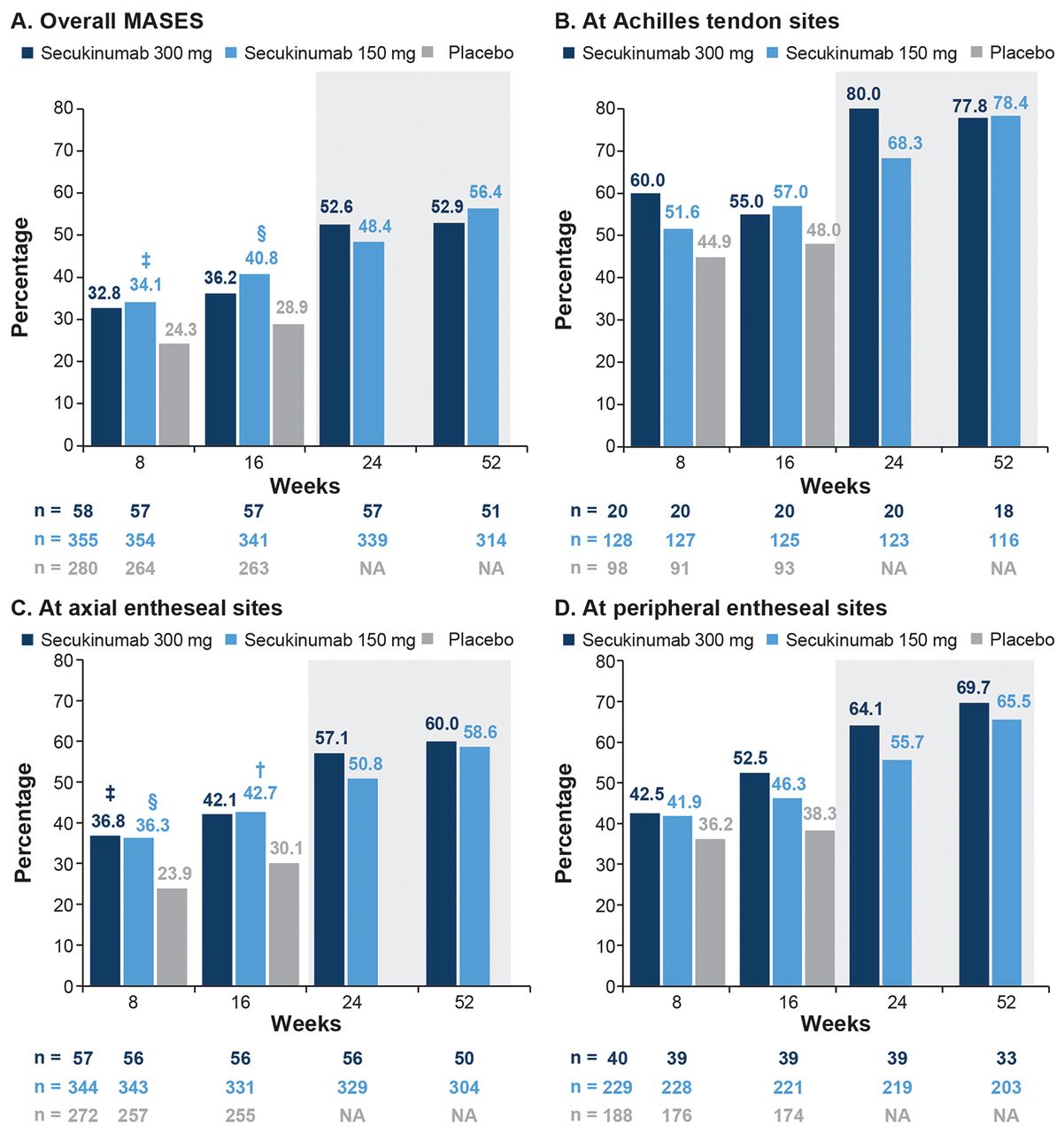
Mean change in MASES score in patients with enthesitis at baseline. At Week 16, the mean change from baseline in MASES score for overall MASES and axial entheseal sites was higher for secukinumab 300 mg (–2.9 [95% CI –1.84 to –0.31; P < 0.01] and –2.9 [95% CI –1.76 to –0.35; P < 0.01], respectively) and 150 mg (–2.4 [95% CI –0.96 to –0.11; P < 0.05] and –2.3 [95% CI –0.89 to –0.10; P < 0.05], respectively) than for PBO (–1.9 and –1.8, respectively). Reductions in MASES score at Achilles tendon and peripheral entheseal sites at Week 16 were –1.0 (95% CI –0.61 to 0.20; not significant [NS]) and –1.6 (95% CI –0.85 to 0.15; NS), respectively, for secukinumab 300 mg; –1.0 (95% CI –0.34 to 0.11; NS) and –1.3 (95% CI –0.42 to 0.15; NS), respectively, for secukinumab 150 mg; and –0.8 and –1.2, respectively, for PBO. Improvements in MASES score were observed through 52 weeks, with the magnitude of response being numerically higher for the overall MASES, axial entheseal sites, peripheral entheseal sites, and Achilles tendon sites with secukinumab 300 mg (–3.9, –3.6, –2.1, and –1.3, respectively) than with secukinumab 150 mg (–3.5, –3.2, –1.9, and –1.2, respectively; Figure 3).

Mean change from baseline in MASES score. § P <0.01; ‡ P <0.05 vs placebo. LS mean and P value are from repeated-measure mixed-effect model until Week 16. Observed data thereafter through Week 52 (shaded area). LS: least squares; MASES: Maastricht Ankylosing Spondylitis Enthesitis Score; NA: not available.
Complete resolution of enthesitis (MASES = 0). At Week 16, 36.2% (secukinumab 300 mg; 95% CI 0.77–2.77; NS) and 40.8% (secukinumab 150 mg; 95% CI 1.27–2.62; P < 0.01 vs PBO) of patients in the 2 secukinumab arms achieved complete resolution of enthesitis for overall MASES, with a corresponding PBO response of 28.9%. At axial entheseal sites, the response rates were 42.1% (secukinumab 300 mg; 95% CI 0.95–3.41; NS), 42.7% (secukinumab 150 mg; 95% CI 1.29–2.69; P < 0.001 vs PBO), and 30.1% (PBO). Response rates for Achilles tendon and peripheral entheseal sites were variable and are shown in 4. At Week 52, 52.9%, 60.0%, 77.8%, and 69.7% patients receiving secukinumab 300 mg and 56.4%, 58.6%, 78.4%, and 65.5% patients receiving secukinumab 150 mg achieved complete resolution of enthesitis for the overall MASES, axial entheseal sites, Achilles tendon sites, and peripheral entheseal sites, respectively (Figure 4).

Proportion of patients with complete resolution of enthesitis (MASES = 0). † P < 0.001; § P < 0.01; ‡ P < 0.05 vs placebo. P values are from logistic regression model. NRI data were used for the analysis up to Week 16, thereafter observed data through Week 52 (shaded area). MASES: Maastricht Ankylosing Spondylitis Enthesitis Score; NA: not available; NRI: nonresponder imputation.
DISCUSSION
Biologics, such as TNFi and IL-17A inhibitors, have been shown to be effective in controlling AS-associated symptoms and are recommended by ASAS and the European Alliance of Associations for Rheumatology for the management of AS.21 Enthesitis, a debilitating characteristic in patients with AS, has been associated with substantial axial and peripheral joint pain.47 Although the importance of enthesitis is acknowledged, the current knowledge on the pathogenesis and treatment of enthesitis is limited.
In various clinical trials, TNFi agents have been shown to improve enthesitis in patients with AS.22,23,24,25,26 However, there remains an unmet medical need for new therapeutic options, particularly in the patient population responding inadequately to TNFi therapy.26–34
Our pooled analysis from a large dataset of patients from the MEASURE studies provides evidence for the efficacy of secukinumab on the resolution of overall enthesitis and at axial entheseal sites in patients with AS through 52 weeks. The most commonly used clinically validated indices for the assessment of enthesitis include MASES and SPARCC.48 In this analysis, we used MASES to assess the resolution of enthesitis. Further, to avoid diluting a treatment effect by including the approximately 30% of patients in the MEASURE studies with no enthesitis at baseline who, therefore, cannot demonstrate improvement on treatment, only patients with MASES > 0 at baseline were included in our analysis to assess improvement over time.
Secukinumab-treated patients with enthesitis at baseline showed greater reductions in MASES, and a greater proportion of patients achieved complete resolution of enthesitis for overall MASES and at axial entheseal sites compared with PBO at Week 16, with improvement trends continuing through Week 52. However, the results for the Achilles tendon and other peripheral entheseal sites did not meet statistical significance for secukinumab vs PBO. Whether these results are due to AS being predominantly an axial disease, the limitations of assessments using MASES, and/or because of higher-than-expected PBO responses requires further research. Secukinumab has demonstrated significant and sustained resolution of enthesitis in patients with PsA,41 as well as rapid and significant improvements in the axial manifestations of PsA.49
The potential limitations of the current analysis include its posthoc nature, as well as the lack of a long-term comparator, since prolonged treatment with PBO is unethical in AS. In turn, the placebo-controlled period of the core MEASURE trials was only up to Week 16. In addition, the number of patients and the mean disease duration were notably lower in the secukinumab 300 mg group than in the 150 mg group, which limits any dose comparison. Further, the conduct of clinical trials is protocol-specified and hence subject to selection bias for inclusion/exclusion of the study population, which may not be fully reflective of real-world clinical experience for the study treatment outcomes. It also cannot be ruled out that some of the patients who did not have baseline enthesitis, and were therefore excluded from the analysis, could have developed enthesitis while on treatment, which would have lowered overall remission rates and increased enthesitis counts. The statistical model for this analysis did not adjust for potential confounding factors such as patients’ sex, which may limit the interpretation of results. In the current study, imaging confirmation of enthesitis was also not performed, nor was this study designed to identify a difference between different regimens of secukinumab administration, which are additional limitations of the analysis.
In conclusion, this posthoc analysis demonstrated that secukinumab 300 mg and 150 mg improved enthesitis assessed by the overall MASES and at axial entheseal sites compared to PBO in patients with AS, extending the evidence for the clinical efficacy of secukinumab in patients with AS that has been reported previously.35,36,37,38 Prospective controlled studies are needed to verify these findings. In addition, the reasons for high PBO responses and variable efficacy at the assessed axial and peripheral entheseal sites warrant further research and highlight the need for a more objective assessment of enthesitis in patients with axSpA.
ACKNOWLEDGMENT
The authors thank the patients who participated in this study; the study investigators; and John Gallagher, Novartis Pharmaceuticals UK Ltd., for valuable review. Manuscript writing support, under the guidance of all authors, was provided by Amit Agarwal and Priyanka Malla, Novartis Healthcare Pvt. Ltd., Hyderabad, India. The first draft of this manuscript was written by Priyanka Malla based on input from all the authors.
Footnotes
This study was funded by Novartis Pharma AG, Basel, Switzerland.
GS has received grant/research support from BMS, Celgene, GSK, Eli Lilly, and Novartis; consultancy fees for AbbVie, BMS, Celgene, Janssen, Eli Lilly, Novartis, and UCB; and speakers bureau fees from AbbVie, BMS, Celgene, Janssen, Eli Lilly, Novartis, and Pfizer. XB has been a consultant for AbbVie, BMS, Chugai, MSD, Novartis, Pfizer, UCB, and received speakers bureau fees from AbbVie, BMS, Chugai, MSD, Novartis, Pfizer, and UCB. FVDB has received consultant/speaker fees from AbbVie, BMS, Celgene, Galapagos, Janssen, Eli Lilly, MSD, Novartis, Pfizer, Sanofi, and UCB. AD has received grant/research support from AbbVie, Eli Lilly, GSK, Novartis, Pfizer, and UCB; and has been a consultant for AbbVie, Amgen, Boheringer Ingelheim, Celgene, Eli Lilly, Gilead, GSK, Janssen, Novartis, Pfizer, and UCB. LG has received grant/research support from Novartis, Pfizer, and UCB, and has been a consultant for AbbVie, Gilead, GSK, Eli Lilly, Novartis, Pfizer, and UCB. MØ has received speaker/consultant fees from AbbVie, BMS, Boehringer-Ingelheim, Celgene, Eli Lilly, Gilead, Hospira, Janssen, Merck, Novartis, Novo, Orion, Pfizer, Regeneron, Roche, and UCB; and research grants from AbbVie, Celgene, Centocor, Merck, and Novartis. ADG is an employee of Novartis. SM, TF, AW, BP, and AS are employees and shareholders of Novartis.
- Accepted for publication February 26, 2021.
- © 2021 The Journal of Rheumatology
REFERENCES
DATA SHARING
The datasets generated and/or analyzed during the current study are not publicly available. Novartis is committed to sharing with qualified external researchers access to patient-level data and supporting clinical documents from eligible studies. These requests are reviewed and approved based on scientific merit. All data provided are anonymized to respect the privacy of patients who have participated in the trial in line with applicable laws and regulations. The data may be requested from the corresponding author of the manuscript.
ONLINE SUPPLEMENT
Supplementary material accompanies the online version of this article.








{kind=link}
{kind=link}
{kind=link}
{kind=link}