

Abstract
Objective. This study evaluated the effect of ixekizumab (IXE) on self-reported functioning and health in patients with radiographic axial spondyloarthritis (r-axSpA) who were either biological disease-modifying antirheumatic drug (bDMARD)–naïve or failed at least 1 tumor necrosis factor inhibitor (TNFi).
Methods. In 2 multicenter, randomized, double-blind, placebo-controlled, and active-controlled (bDMARD-naïve only) trials, patients with r-axSpA were randomly assigned to receive 80 mg of IXE [every 2 weeks (Q2W) or every 4 weeks (Q4W)], placebo (PBO), or adalimumab (ADA; bDMARD-naïve only). After 16 weeks, patients who received PBO or ADA were rerandomized to receive IXE (Q2W or Q4W) up to Week 52. Functioning and health were measured by the generic 36-item Short Form Health Survey (SF-36) and the disease-specific Assessment of Spondyloarthritis international Society Health Index (ASAS HI). Societal health utility was assessed by the 5-level EuroQol-5 Dimension (EQ-5D-5L).
Results. At Week 16, both doses of IXE in bDMARD-naïve and TNFi-experienced patients resulted in larger improvement in SF-36, ASAS HI, and EQ-5D-5L versus placebo. For SF-36, the largest improvements were seen for the domains of bodily pain, physical function, and role physical. A larger proportion of patients reaching improvement in ASAS HI ≥ 3 as well as an achievement of ASAS HI good health status was reported in patients treated with IXE. Improvements were maintained through Week 52.
Conclusion. IXE significantly improved functioning and health as assessed by both generic and disease-specific measures, as well as societal health utility values in patients with r-axSpA, as measured by SF-36, ASAS HI, and EQ-5D-5L at Week 16, and improvements were sustained through 52 weeks.
Radiographic axial spondyloarthritis (r-axSpA), also referred to as ankylosing spondylitis (AS), is a potentially disabling chronic inflammatory disease of the axial skeleton that affects 0.2–0.5% of the population1,2,3,4,5. R-axSpA is characterized by inflammation and new bone formation in the sacroiliac joints and spine6. Patients with r-axSpA present diverse clinical features including inflammatory back pain, limited physical function and activities (e.g., standing, walking, reaching), stiffness, fatigue, impaired mental function (e.g., depression, anxiety), and restricted social relationships, all of which contribute to reduced overall functioning and health6,7,8,9. Measures that assess the integrated effect of this broad range of different impairments into 1 instrument are referred to as overall health or health-related quality of life (HRQOL) measures, and provide insight into how the disease actually alters the daily life of patients. Therefore, overall health or HRQOL is an important outcome measure when assessing the efficacy of treatments.
Current treatments for the management of r-axSpA include nonpharmacological management such as physical therapy and education, as well as pharmaceutical treatment. Nonsteroidal antiinflammatory drugs (NSAID) are recommended as first-line treatments for improving back pain and stiffness1,10. Biologic disease-modifying antirheumatic drugs (bDMARD) such as tumor necrosis factor inhibitors (TNFi) are recommended when NSAID fail10. Treatment with TNFi such as etanercept, infliximab, adalimumab (ADA), golimumab, and certolizumab pegol have demonstrated high efficacy on disease activity and improvement of 36-item Short Form Health Survey (SF-36) scores1,3. However, approximatively 40% of patients with r-axSpA still report high disease activity despite availability of multiple TNFi11. Consequently, there is a need for alternative treatment options in patients with r-axSpA who do not respond to, or do not tolerate, TNFi11,12.
Recently, growing evidence indicates the interleukin (IL)-17 pathway, and in particular IL-17A, plays a critical role in r-axSpA pathogenesis13,14. Ixekizumab (IXE) is an IgG4 monoclonal antibody that selectively targets IL-17A with very high affinity15,16. The U.S. Food and Drug Administration (FDA) and the European Medicines Agency approved IXE for the treatment of moderate-to-severe plaque psoriasis and active psoriatic arthritis in adults, and for adult patients with AS. Recently, IXE was approved by the FDA for moderate-to-severe pediatric psoriasis. Results from 2 completed phase III randomized, double-blind, placebo-controlled trials demonstrated IXE 16-week and 52-week treatment efficacy in bDMARD-naïve (COAST-V) and TNFi-experienced (COAST-W) patients with AS17,18,19. Previously, we reported statistically significant improvement versus placebo (PBO) at Week 16 of the HRQOL endpoints measured by the mean changes in SF-36 (both IXE doses), and the Assessment of Spondyloarthritis international Society Health Index (ASAS HI; COAST-V, both IXE doses; COAST-W, IXE Q4W only) in bDMARD-naïve and TNFi-experienced patients18,19. In these studies, improvements were sustained through Week 5217. In the present study, in addition to improvements of IXE by SF-36 and ASAS HI means through Week 52 in patients with active r-axSpA TNFi nonresponders, we report the SF-36 domains, the proportion of patients with improvement in ASAS HI ≥ 3 from baseline, the proportion of patients achieving an ASAS HI good health status (ASAS HI ≤ 5), and 5 level-EuroQol-5D (EQ-5D-5L), through 52 weeks.
MATERIALS AND METHODS
Studies designs. COAST-V (ClinicalTrials.gov: NCT02696785) and COAST-W (ClinicalTrials.gov: NCT02696798) are phase III, multicenter, active (COAST-V only), and PBO randomized controlled trials (RCT) with a 52-week duration, evaluating the efficacy and safety of IXE in patients with r-axSpA. The main ethics committee was Schulman Associates IRB, Cincinnati, Ohio, USA (IRB# 201506061 for COAST-V, and 201506079 for COAST-W). The full lists of investigators and sites are provided in the primary manuscript supplements18,19. Patient enrollment and data collection occurred at 84 sites in 12 countries in the COAST-V trial, and in 106 sites in 15 countries in the COAST-W trial. The studies were approved by the ethical review boards at each participating site before the study start. The RCT conform with Good Clinical Practices, International Council for Harmonization, and local laws and regulations, and were conducted in accordance with the Declaration of Helsinki principles. All enrolled patients provided written informed consent before participating in the trials.
Participants. Inclusion criteria have been previously detailed18,19. Briefly, eligible patients were ≥ 18 years with an established diagnosis of r-axSpA and fulfilling ASAS criteria (sacroiliitis on radiograph by modified New York criteria and at least 1 SpA feature). The sacroiliac joint radiograph reading was performed centrally by 2 independent readers, with adjudication if necessary. Participants in COAST-V were bDMARD-naïve, whereas in the COAST-W trial, participants had failed between 1 and 2 TNFi prior to enrollment.
Interventions. The 16-week results of the COAST-V and COAST-W interventions have been previously described18,19. In COAST-V, patients were randomly assigned using a 1:1:1:1 ratio to IXE 80 mg every 2 weeks (Q2W), IXE 80 mg every 4 weeks (Q4W), ADA 40 mg Q2W, or PBO. In COAST-W, patients were randomly assigned using a 1:1:1 ratio to IXE Q2W, IXE Q4W, or PBO. In both trials, participants initially assigned to IXE treatment were randomly assigned in a 1:1 ratio to receive a starting dose of either 80 mg IXE or 160 mg IXE (two 80-mg injections) for the first dose at Week 0. Patients completing Week 16 entered a double-blind extended treatment period (ETP; Weeks 16–52). During this period, patients originally randomized to PBO or ADA (COAST-V only) were rerandomized 1:1 to IXE Q2W or IXE Q4W (160 mg starting dose for patients switching from PBO; 80 mg starting dose for patients switching from ADA). Patients originally randomized to IXE Q2W or IXE Q4W continued these regimens. All doses were administrated subcutaneously using masked prefilled manual syringes.
Self-reported functioning and health as assessed by generic and disease-specific measures. The effects of IXE on HRQOL were assessed using 2 secondary major endpoints, the SF-36 and ASAS HI. Assessments were recorded at Week 0 (baseline), 4, 8, 16, 36, and 52. SF-36 is a 36-item patient-administered measure designed as a short, generic assessment of HRQOL including the following domains: physical and social functioning, physical and emotional roles, bodily pain, general health, vitality, and mental health. The domain scores range from 0 to 100, with higher scores indicating better levels of function and/or better health. The physical component summary (PCS) and mental component summary (MCS) scores are calculated based on differential weighting of the 8 domains having been normalized to t scores. Items were answered based on Likert scales of 3–5. SF-36 version 2 (acute version), which utilizes a 1-week recall period, was used in the COAST-V and COAST-W studies20.The scaled scores (0–100) were used in the spydergrams21, and the least square mean (LSM) changes from baseline in t scores were cited in Table 1. The 1998 norms were used in previous publications, reporting SF-36 values for Week 0–16 described in this manuscript18, 19, so the current data for Weeks 0–16 are analyzed with this norm for consistency. The data after Week 16 have since been analyzed using the updated 2009 norms, which are used in the latest version of the SF-36 manual. The 1998 and 2009 norms are minimally different. The calculation of age- and sex-matched norms for each domain in the spydergram in Figure 1 is based on 1998 US population norms and matched for the age and sex distribution of the protocol population.
Baseline demographics and disease characteristics, COAST-V and COAST-W (intent-to-treat population).

SF-36 physical component summary scores change from baseline COAST-V and COAST-W (intent-to-treat population). (A–B) Comparisons with PBO were made using a mixed effects model for repeated measures up to Week 16 (least squares means). (C–F) Descriptive statistics were provided using mBOCF for missing data imputation approach. Week 0–16 data are based on 1998 general US population (norm 1998) as norms (A–B), and reports after Week 16 are based on 2009 general US population (norm 2009) as norms (C–F). ** P < 0.01. *** P < 0.001. ADA: adalimumab 40 mg every 2 weeks; bDMARD: biologic disease-modifying antirheumatic drugs; IXE: ixekizumab; Q2W: every 2 weeks; Q4W: every 4 weeks; mBOCF: modified baseline observation carried forward; PBO: placebo; SF-36: 36-item Short Form Health Survey; TNFi: tumor necrosis factor inhibitor.
The ASAS HI is a disease-specific health index designed to assess effect of the disease on patients and covers areas of physical, emotional, and social functioning. This 17-item instrument has sum scores ranging from 0 (good health) to 17 (poor health)22. The clinically meaningful change is defined as a difference of ≥ 3 points between 2 timepoints, and a good health status is defined by a score ≤ 5 points at 1 timepoint23,24.
The EQ5D5L provides societal preferences for health states (health utilities) based on 5 dimensions of health: mobility, self-care, usual activities, pain/discomfort, and anxiety/depression. The patient-reported EQ-5D-5L descriptive system was converted into a societal utility value using the available UK population-based algorithm to produce a patient-level index score between –0.59 and 1.0 (continuous variable)25.
Statistical analyses. Analyses were conducted on the intent-to-treat (ITT) population for patients initially randomized to IXE (Weeks 0–52), ADA, or PBO (Weeks 0–16). The analyses of the ETP (Weeks 16–52) for patients initially assigned to ADA or PBO were conducted. For comparisons between each IXE treatment group (Q2W or Q4W) and PBO up to Week 16, the primary analysis method for continuous outcomes (SF-36 domains and component scores, ASAS HI, and EQ-5D-5L) was a mixed effects model for repeated measures (MMRM) with treatment, geographic region, baseline C-reactive protein (CRP) status (nonelevated or elevated; elevated defined as > 5.00 mg/L), number of prior anti-TNFi used (COAST-W only), baseline value, visit, baseline value-by-visit, and treatment-by-visit interaction as fixed factors. Treatment comparisons for categorical outcomes (improvement in ASAS HI ≥ 3 points obtained, and ASAS HI good health status achieved) were performed using logistic regression with treatment, geographic region, baseline CRP status (nonelevated or elevated; elevated defined as > 5.00 mg/L), and the number of prior anti-TNFi used (COAST-W only) in the model. For the ETP (Weeks 16–52), no treatment group comparisons were conducted. For SF-36 outcomes and EQ-5D-5L, no imputation for missing data was done when using MMRM modeling up to Week 16, while descriptive statistics were provided for patients initially randomized to IXE (Weeks 0–52) and for the ETP population using the modified baseline observation carried forward imputation approach for missing data. For categorical ASAS HI outcomes, missing data were imputed as improvement < 3 points and ASAS HI > 5 using nonresponder imputation. The statistical analyses were performed using SAS software version 9.3 or higher (SAS Institute Inc.).
RESULTS
Of the 341 (COAST-V) and 316 (COAST-W) patients included in this analysis, sample sizes for COAST-V were n = 87 (PBO), n = 81 (IXE Q4W), n = 83 (IXE Q2W), and n = 90 (ADA); and for COAST-W were n = 104 (PBO), n = 114 (IXE Q4W), and n = 98 (IXE Q2W; Table 1). Sample sizes were balanced between treatment groups. Demographics and baseline clinical characteristics for the ETP populations were similar between treatment groups within each study (Table 1) and similar to those in the ITT populations18,19. SF-36, ASAS HI, and EQ5D5L baselines were also balanced between treatment arms within each trial.
IXE improves functioning and health as assessed by generic measure SF-36. Improvements in SF-36 PCS for IXE versus PBO were significantly larger throughout the 16 weeks assessed (Figures 1A,B). Improvements in the PCS scores with IXE were consistent between bDMARD-naïve and TNFi-experienced patients, with significant improvements reported as early as Week 4. Both IXE dose groups showed sustained improvement on the SF-36 PCS through Week 52 (Figures 1C,D). Patients who were bDMARD-naïve treated with the active reference ADA also showed significant improvement in PCS treatment response score versus PBO up to 16 weeks (Figure 1A). Interestingly, patients treated with ADA and rerandomized at Week 16 to IXE demonstrated continued improvement in the PCS, and reached a similar level at Week 52 compared with patients who received IXE from Week 0 (Figure 1E). Patients initially assigned to the PBO arm and who received IXE starting at Week 16 reported a rapid improvement throughout the ETP (Figures 1E,F). In the bDMARD-naïve patients, nonsignificant differences between groups in the improvements of MCS were observed (Supplementary Figure 1, available with the online version of this article). Statistically significant improvements in the MCS were reported at Week 4 (IXE Q4W only), and Week 8 in TNFi-experienced patients.
The effects of IXE on the SF-36 domains at Week 16 and Week 52 compared with baseline in the bDMARD-naïve and TNFi-experienced patients are shown in Figure 2. Improvements in all SF-36 domains were reported up to Week 52 in bDMARD-naïve and TNFi-experienced patients treated with IXE. Both bDMARD-naïve and TNFi-experienced patients treated with IXE reported larger improvements compared with PBO in SF-36 domains at Week 16 and sustained benefits through Week 52. By Week 52, the largest improvements (scaled score) among patients treated with IXE were observed in the bodily pain and physical functioning category (bDMARD-naïve; +24.7 points from baseline for Q4W, + 23.5 for Q2W, +18.0 for Q4W, +20.7 for Q2W, respectively) and TNFi-experienced patients (+22.1 points from baseline for Q4W, +21.3 for Q2W; +15.9 for Q4W, and +19.6 for Q2W, respectively). Patients treated with the active reference ADA also showed consistent improvement in all SF-36 domains throughout the 16 weeks assessed in the blinded treatment dosing period.
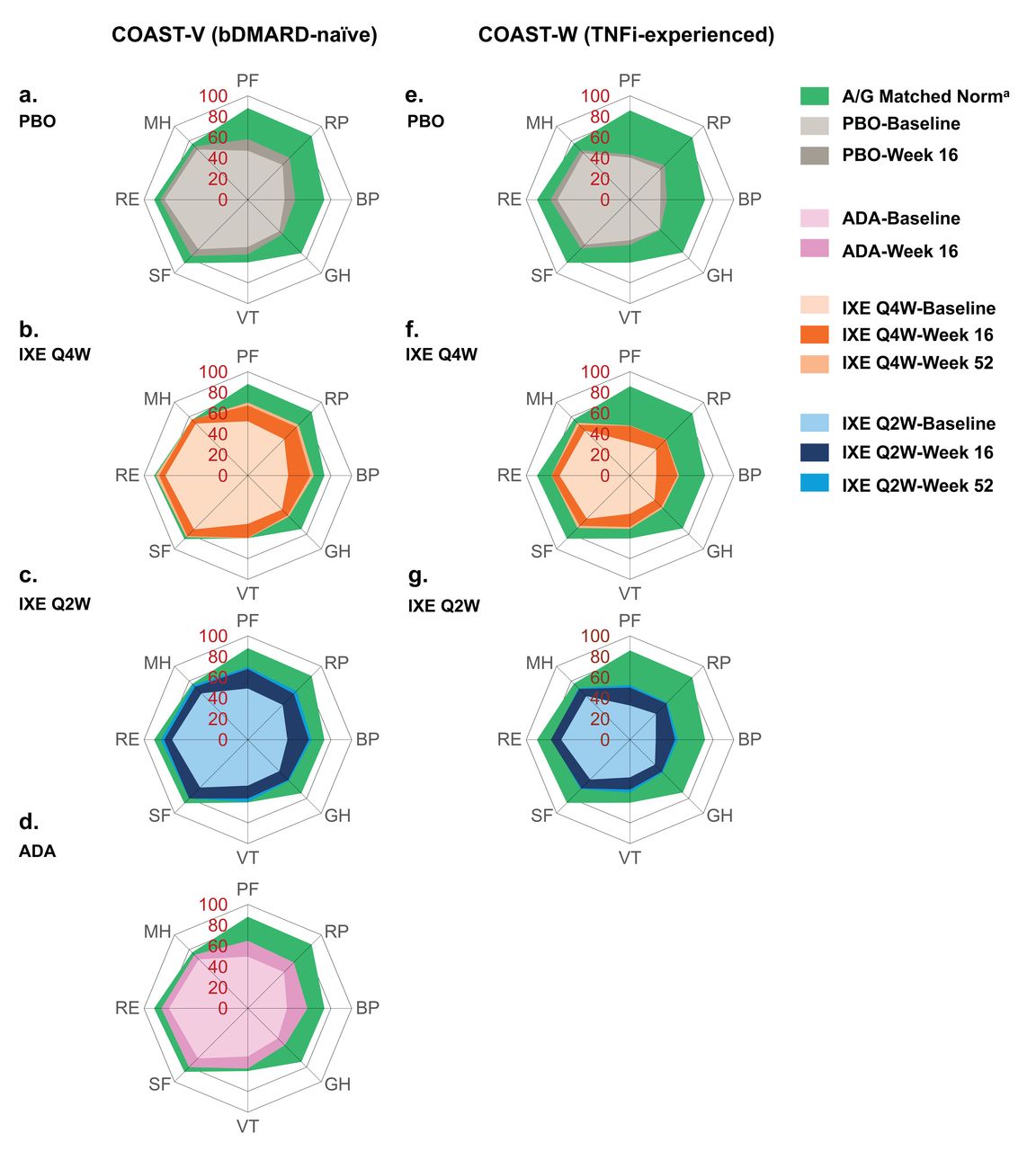
SF-36 domain scores at baseline, and 16 and 52 weeks, COAST-V and COAST-W (intent-to-treat population). The spydergrams depict mBOCF SF-36 domain scores (scale 0–100) and US A/G-matched normative values. SF-36 A/G-matched norms are based on 1998 US population norms and patient counts for each age and gender distribution of the protocol population. a1998 US population. ADA: adalimumab 40 mg every 2 weeks; A/G: age/gender; BP: bodily pain; bDMARD: biologic disease-modifying antirheumatic drugs; GH: general health; IXE: ixekizumab; IXE Q2W: 80 mg ixekizumab every 2 weeks; IXE Q4W: 80 mg ixekizumab every 4 weeks; MH: mental health; mBOCF: modified baseline observation carried forward; PBO: placebo; PF: physical functioning; RE: role emotional; RP: role physical; SF: social functioning; SF-36: 36-item Short Form Health Survey; TNFi: tumor necrosis factor inhibitors; VT: vitality.
Actual scores of SF-36 domains and components at baseline, and mean changes at Weeks 16 and 52 for the bDMARD-naïve and TNFi-experienced patients are presented in Supplementary Table 1 (available with the online version of this article). In general, bDMARD-naïve patients reported numerically higher numbers for all SF36 baseline measures compared with TNFi-experienced patients, indicating better functioning health. Significant improvement of some SF-36 domains was already observed at the first assessment (Week 4; data not shown).
IXE improves functioning and health measured by the disease-specific ASAS HI. At Week 16, bDMARD-naïve patients receiving IXE reported a significantly larger improvement from baseline on ASAS HI versus PBO [–2.36 for Q4W (P = 0.01), –2.74 for Q2W (P < 0.001) vs –1.25 for PBO; Figure 3A]. These improvements with IXE treatment were seen as early as Week 4, remained higher than PBO through Week 16, and sustained through Week 52. IXE Q4W bDMARD-naïve patients achieved numerically similar ASAS HI mean change from baseline as patients who received IXE Q2W (–2.7 vs –3.3 at Week 52; Figure 3C). Patients treated with the active reference ADA also showed consistent significant improvement in ASAS HI mean change from baseline throughout the 16 weeks assessed (Figure 3A). Patients who received ADA or PBO during the blinded treatment dosing period and switched to IXE at Week 16 demonstrated continued numeric improvements in ASAS HI through Week 52 (Figure 3E). Both IXE regimens (Q2W and Q4W) sustained similar improvements through Week 52. Patients in the bDMARD-naïve arm experienced a numerically greater improvement of ASAS HI mean change versus TNFi-experienced patients when treated with IXE Q4W (–2.4 vs –1.9 at Week 16 and –2.7 vs –2.3 at Week 52) or IXE Q2W (–2.7 vs –1.6 at Week 16 and –3.3 vs –2.5 at Week 52; Figures 3A–D).

ASAS HI least squares mean change from baseline COAST-V and COAST-W (intent-to-treat population). (A–B) Comparisons with PBO were made using a mixed effects model for repeated measures up to Week 16. (C–F) Descriptive statistics were provided for Weeks 36–52 using mBOCF for missing data imputation approach. *P < 0.05. **P < 0.01. ***P < 0.001. ADA: adalimumab 40 mg every 2 weeks; ASAS HI: Assessment of Spondyloarthritis international Society Health Index; bDMARD: biologic disease-modifying antirheumatic drug; IXE: ixekizumab; IXE Q2W: IXE dosed every 2 weeks; IXE Q4W: IXE dosed every 4 weeks; mBOCF: modified baseline observation carried forward; PBO: placebo; TNFi: tumor necrosis factor inhibitors.
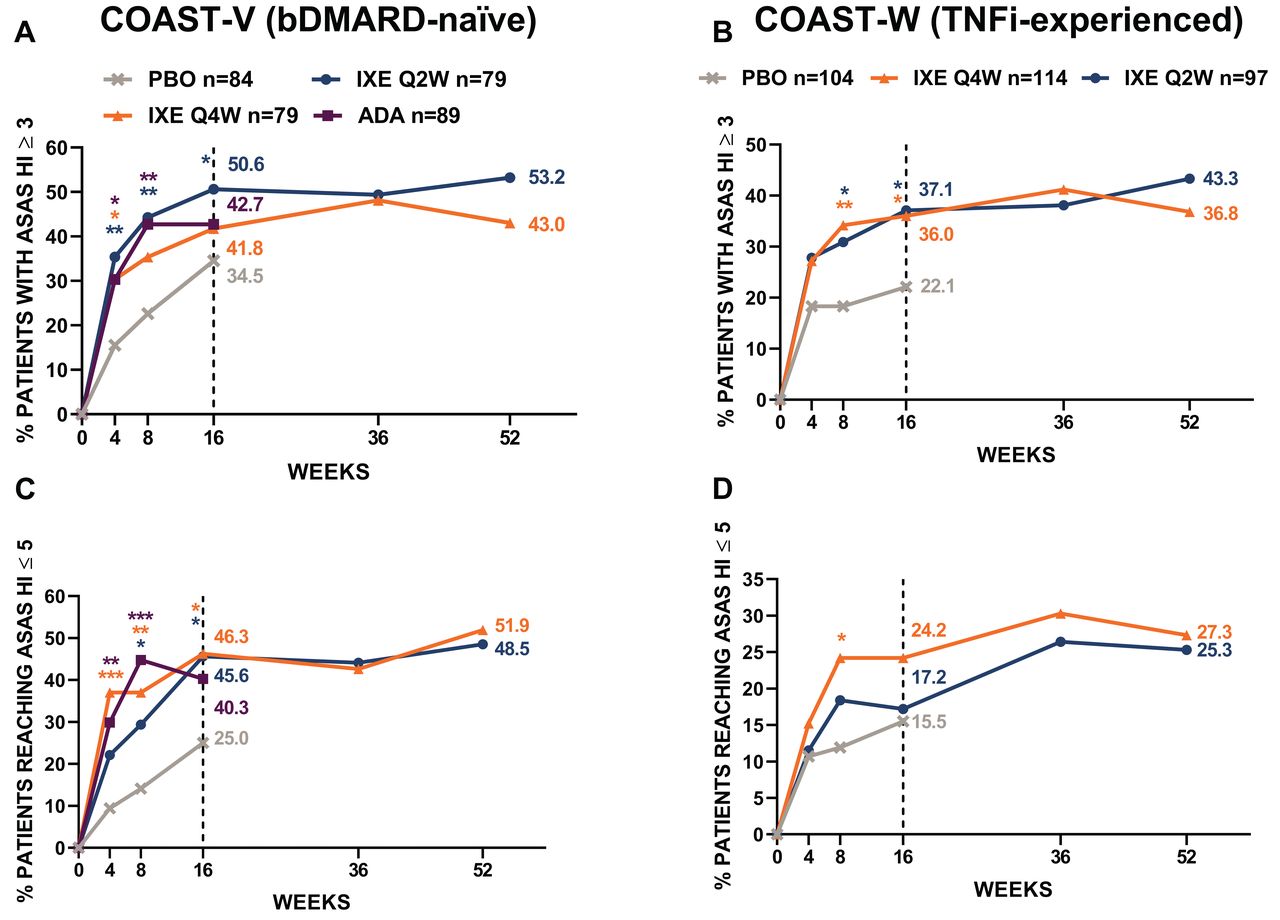
The proportion of patients achieving an improvement in ASAS HI ≥ 3 points change from baseline were also analyzed (Figure 4). At baseline, the proportion of bDMARD-naïve patients with ASAS HI ≥ 3 ranged from 95.2% to 98.9%, and from 99.0% to 100% among TNFi-experienced patients. Compared with PBO, the improvement in ASAS HI ≥ 3 at Week 16 was achieved by a higher proportion of bDMARD-naïve patients treated with IXE Q4W (34.5% vs 41.8%, P = 0.31) and a significantly higher proportion treated with Q2W (34.5% vs 50.6%, P = 0.033), and improvements were consistent through Week 52 (Q4W 43.0% and Q2W 53.2%). The proportion of patients treated with IXE achieving improvement in ASAS HI ≥ 3 throughout the 52 weeks were 53.2% for Q2W and 43.0% for Q4W (bDMARD-naïve), and 43.3% and 36.8% (TNFi-experienced; Figures 4A,B). The proportion of patients achieving improvement in ASAS HI ≥ 3 in the ADA arm was also significantly greater than the PBO arm (Figure 4A). TNFi-experienced patients achieved ASAS HI ≥ 3 more often than those on PBO, with significant differences at Week 16 (22.1% for PBO vs 37.1% for IXE Q2W, P = 0.032, and 36.0% for IXE Q4W, P = 0.026). At Week 52, 43.3% of IXE Q2W and 36.8% of IXE Q4W TNFi-experienced patients achieved an improvement in ASAS HI ≥ 3 (Figure 4B).

Proportion of patients with ASAS HI improvement ≥ 3 and achieving ASAS HI ≤ 5 (good health status) COAST-V and COAST-W (intent-to-treat population). Missing data were imputed using nonresponder imputation. Comparisons with PBO were made using logistic regression model up to Week 16. Descriptive statistics were provided for Weeks 36–52. *P < 0.05. **P < 0.01. ***P < 0.001. ADA: adalimumab 40 mg every 2 weeks; ASAS HI: Assessment of Spondyloarthritis international Society Health Index; bDMARD: biologic disease-modifying antirheumatic drug; IXE: ixekizumab; IXE Q2W: IXE dosed every 2 weeks; IXE Q4W: IXE dosed every 4 weeks; mBOCF: modified baseline observation carried forward; PBO: placebo; TNFi: tumor necrosis factor inhibitors.
At baseline, the proportion of patients with no good health status ranged from 66.7% to 81.9% in bDMARD-naïve patients and from 80.8% to 88.8% in TNFi-experienced patients (Table 1). In general, numerically similar improvements in the proportion of patients reaching good health status were reported in both IXE dose groups (Figures 4C,D). At Week 16, good health status was achieved by 46.3% and 45.6% of the bDMARD-naïve patients treated with IXE Q4W and Q2W, respectively, compared with PBO (25.0%, P < 0.05 for both doses). Good health status was achieved by 24.2% and 17.2% of the TNFi-experienced patients treated with IXE Q4W and Q2W, respectively, compared with PBO (15.5%) at Week 16. Also, at Week 16, 40.3% of bDMARD-naïve patients who received ADA reached good health status (Figure 4C). The proportion of patients achieving good health status was sustained through 52 weeks, with 51.9% and 48.5% of bDMARD-naïve patients treated with IXE Q4W and Q2W, respectively, and by 27.3% and 25.3% of TNFi-experienced patients treated with IXE Q4W and Q2W, respectively (Figures 4C,D).
IXE improves health utility assessed by EQ-5D-5L. The results for EQ-5D-5L health utilities are provided in Figure 5. Each IXE treatment group compared to the PBO group had significantly larger improvements at Week 16 in both bDMARD-naïve (0.19 for IXE Q4W and 0.19 for IXE Q2W vs 0.10 for PBO) and TNFi-experienced patients (0.16 for IXE Q4W and 0.16 for IXE Q2W vs 0.08 for PBO). Additionally, the active reference ADA treatment group had a significantly greater proportion of patients with improvements in EQ-5D-5L at Week 16 compared with PBO. Effects were sustained throughout Week 52 in both IXE treatment groups (bDMARD-naïve: 0.18 Q4W, 0.20 Q2W; TNFi-experienced: 0.21 Q4W, 0.20 Q2W). Patients who received ADA and were switched to IXE demonstrated continued numeric improvements in EQ-5D-5L (from 0.16 at Week 16, to 0.20 at Week 52). All patients who received PBO from Week 0 to 16 and switched to IXE showed rapid improvements and reached a similar score at Week 52 as patients who received IXE from Week 0. In bDMARD-naïve patients, this improvement was 0.20 for PBO/IXE versus 0.18 IXE Q4W, and 0.20 for IXE Q2W, and in TNFi-experienced patients this improvement was 0.19 for PBO/IXE versus 0.21 for IXE Q4W, and 0.20 for IXE Q2W.
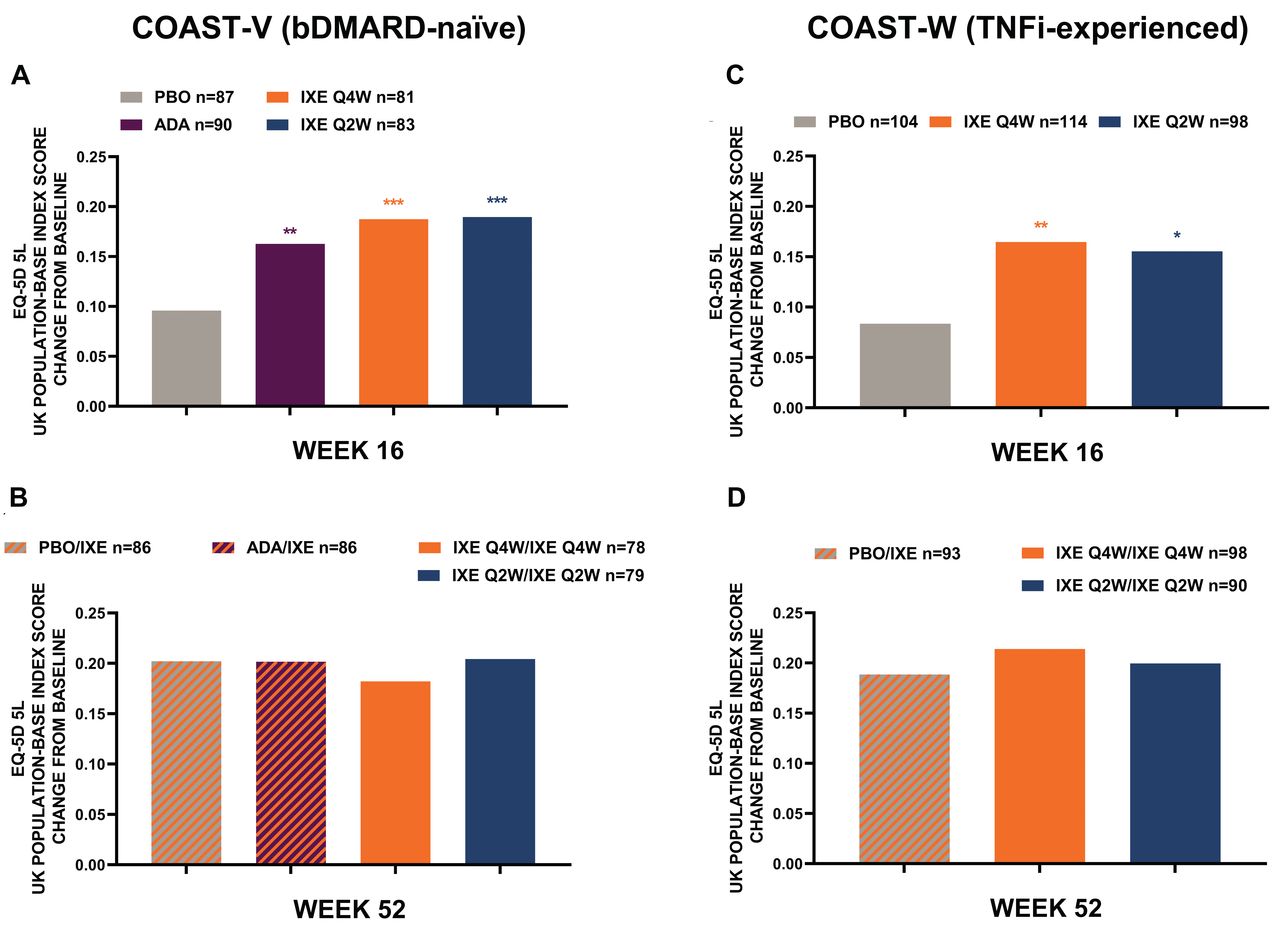
EQ-5D-5L UK population index score least-squares mean change from baseline COAST-V and COAST-W. Intent-to-treat population was used at Week 16, and extended treatment period population at Week 52. Missing data were imputed using nonresponder imputation. Comparisons with PBO were made using logistic regression model at Week 16. Descriptive statistics were provided at Week 52 using mBOCF for missing data imputation approach. *P < 0.05. **P < 0.01. ***P < 0.001. ADA: adalimumab 40 mg every 2 weeks; bDMARD: biologic disease-modifying antirheumatic drugs; EQ-5D-5L: 5-level EuroQol-5 Dimension; IXE: ixekizumab; IXE Q2W: IXE dosed every 2 weeks; IXE Q4W: IXE dosed every 4 weeks; mBOCF: modified baseline observation carried forward; PBO: placebo; TNFi: tumor necrosis factor inhibitors.
DISCUSSION
In the present analysis, we demonstrate that the IL-17A inhibitor IXE significantly improved self-reported functioning and health as well as societal health utilities through Week 0–16 among both bDMARD-naïve and TNFi-experienced patients with active r-axSpA, and improvements were sustained through Weeks 16 to 52. Significant improvements compared with PBO were observed at Week 16 in bDMARD-naïve patients treated with both IXE Q4W or IXE Q2W for all outcomes (except for ASAS HI ≥ 3 with IXE Q4W). In these patients, improvements were observed as early as the first assessment at Week 4 for mean change from baseline in SF-36 PCS and ASAS HI, as well as a proportion experiencing a meaningful improvement in ASAS HI or reaching a “good” ASAS HI (IXE Q4W only). Similarly, TNFi-experienced patients treated with IXE reported a significant improvement versus PBO at Week 16 for societal health utility values as well as most generic and disease-specific measures of function and health outcomes, except the proportion of patients reaching a good ASAS HI ≤ 5, where nonsignificant differences in the advantage of the IXE-treated patients were observed. At baseline, SF-36 MCS was within the normal range; therefore, ranges of improvement were limited in both bDMARD-naïve and TNFi-experienced patients. There was no meaningful difference in responses based on IXE dosing regimen (Q2W or Q4W).
Patients in the bDMARD-naïve arm had a numerically higher response to IXE treatment compared with TNFi-experienced patients; however, statistical analysis comparing these groups has not been conducted. At baseline, the duration of symptoms since the onset of r-axSpA was higher in the TNFi-experienced patients versus the bDMARD-naïve patients (18.4 vs 16.0 yrs on average among the arms). These data could indicate bDMARD-naïve patients may have more opportunity for improvement because they have more reversible physical impairment. These data could also indicate axial pain reported by TNFi-experienced patients may partly have sources other than inflammation. Further analyses that aim to investigate inflammation outcomes could be conducted to test this hypothesis.
The improvements in overall health or HRQOL outcomes observed in patients with r-axSpA in this 52-week placebo-controlled trial are consistent with the SF-36 and/or EQ-5D results from the phase III placebo-controlled studies with the other IL-17A inhibitor secukinumab26 or anti-TNF agents27,28,29. However, COAST-V and COAST-W were the first trials to report ASAS HI outcome to assess disease-specific functioning and health. Due to difference in patient population and study design, a direct comparison between studies and agents is challenging, even when analysis on individual data would be performed as contextual factors, which are relevant for appraisal of self-reported overall health that are usually not measured in trials. Despite the tremendous interest to evaluate the efficacy and safety of IXE through 52 weeks, the design of the EPT (Weeks 16–52) presents some limitations. The interpretation of data in an extended treatment period without a control arm (PBO) is challenging. Therefore, the long-term superiority of IXE versus PBO from Weeks 16 to 52 cannot be established. The main strength of the present analysis is the comparison of 2 separate trials with 2 independent populations of patients. This combined analysis provides valuable information regarding the efficacy of IXE on self-reported functioning and health outcomes in both bDMARD-naïve and TNFi-experienced patients.
In conclusion, the present analyses demonstrate IXE significantly improved functioning and health outcomes (as assessed by generic and disease-specific measures) as well as societal health utility values as early as Week 4, and sustained through Week 52 among patients with r-axSpA who are bDMARD-naïve or have had a prior inadequate response or intolerance to TNFi.
ACKNOWLEDGMENT
Elsa Mevel, PhD, and Dana Schamberger, MA, provided writing and editorial assistance. The authors thank the study participants, caregivers, and investigators.
Footnotes
Sponsorship for this study and article processing charges were funded by Eli Lilly and Company. All authors had full access to all the data in this study and take complete responsibility for the integrity of the data and accuracy of the data analysis.
UK has received grant/research support and consultancy fees from AbbVie, Chugai, Eli Lilly and Company, Grünenthal, Janssen, MSD, Novartis, Pfizer, Roche, and UCB. JCW has received consultant and/or speaking fees from Johnson & Johnson, AbbVie, UCB, and Novartis. DvdH is a consultant for AbbVie, Amgen, Astellas, AstraZeneca, BMS, Boehringer Ingelheim, Celgene, Daiichi Sankyo, Eli Lilly and Company, Galapagos, Gilead, Janssen, Merck, Novartis, Pfizer, Regeneron, Roche, Sanofi, and UCB. FvdB has received research grant support, consultancy honoraria, or speaker fees from AbbVie, Eli Lilly and Company, Janssen, Merck, and UCB Pharma, and received consultancy honoraria or speaker fees from Bristol Myers-Squibb, Celgene, Novartis, Pfizer, and Sanofi. JAW has received grants and/or consultant fees from AbbVie, Celgene, Novartis, Pfizer, UCB, and Eli Lilly and Company. AB has received consulting fees from Eli Lilly and Company, Novartis, and UBC, has received grant support from AbbVie and Celgene. LSG has received grant and research support from AbbVie, Amgen, Novartis, and UCB, and has received consulting fees from Eli Lilly and Company, Galapagos, Janssen, Novartis, and Pfizer. TH, HC, YD, XL, and RB are current employees and shareholders of Eli Lilly and Company. VS reports consulting fees from Eli Lilly and Company, AbbVie, Amgen, Anthera, AstraZeneca, Bristol Myers Squibb, Boehringer Ingelheim, Celltrion, EMD Serono, Genentech/Roche, GlaxoSmithKline, Janssen, Novartis, Pfizer, Regeneron, Sanofi, and UCB. JB is a consultant for AbbVie, Amgen, Celgene, Eli Lilly and Company, Janssen, MSD, Novartis, Pfizer, Sanofi, and UCB.
Full Release Article. For details see Reprints and Permissions at jrheum.org.
- Accepted for publication July 6, 2020.
- Copyright © 2021 by the Journal of Rheumatology
Free online via JRheum Full Release option
REFERENCES
DATA AVAILABILITY
The datasets generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request.
ONLINE SUPPLEMENT
Supplementary material accompanies the online version of this article.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}