

Abstract
Objective. The Outcome Measures in Rheumatology Soluble Biomarker Working Group initiated an international, multicenter, prospective study, the Rheumatoid Arthritis (RA) BIODAM cohort, to generate resources for the clinical validation of candidate biomarkers predictive of radiographic progression. This first report describes the cohort, clinical outcomes, and radiographic findings.
Methods. Patients with RA from 38 sites in 10 countries starting or changing conventional synthetic disease-modifying antirheumatic drugs and/or starting tumor necrosis factor inhibitors were followed for 2 years. Participating physicians were required to adhere to a treat-to-target strategy. Biosamples (serum, urine) were acquired every 3 months, radiography of hands and feet every 6 months, and ultrasound of hands and feet every 3 months in a subset. Primary endpoint was radiographic progression by the Sharp/van der Heijde score.
Results. A total of 571 patients were recruited and 439 (76.9%) completed 2-year followup. At baseline, the majority was female (76%), mean age 55.7 years, and mean disease duration 6.5 years. Patients had a mean of 8.4 swollen and 13.6 tender joints, 44-joint count Disease Activity Score (DAS44) 3.8, 77.7% rheumatoid factor–positive or anticitrullinated protein antibody–positive. Percentage of patients in DAS and American College of Rheumatology remission at 2 years was 52.2% and 27.1%, respectively. Percentage of patients with radiographic progression (> 0.5) at 1 and 2 years was 38.2% and 59.9%, respectively.
Conclusion. The RA BIODAM prospective study succeeded in generating an extensive list of clinical, imaging (2343 radiographs), and biosample (4638 sera) resources that will be made available to expedite the identification and validation of biomarkers for radiographic damage endpoints. (Clinicaltrials.gov: NCT01476956, clinicaltrials.gov/ct2/show/NCT01476956)
At the Outcome Measures in Rheumatology (OMERACT) 8 meeting (2006), an international special interest group was assembled to develop validation criteria for a soluble biomarker to substitute for radiographic outcome measures in clinical trials of rheumatoid arthritis (RA), psoriatic arthritis (PsA), and axial spondyloarthritis (axSpA)1,2. At the OMERACT 9 meeting, there was a reappraisal of the OMERACT 8 criteria and an international consensus was generated on a final set of criteria that focused on the performance characteristics of biomarker assays, the importance of addressing potential confounders, and the essential requirement for clinical validation studies3,4. In addition, the group formulated a levels of evidence scheme and a study design template aimed at guiding the conduct of clinical validation studies for soluble biomarkers proposed to replace the measurement of damage endpoints in RA, PsA, and AS5. This prioritization for clinical validation of biomarkers also reflected the international consensus that there was a major unmet need for a modifiable prognostic biomarker that could influence the routine management of these diseases. In particular, prognostic risk prediction tools for damage endpoints based on clinical and laboratory variables currently used in practice lack sufficient predictive capacity and clinical utility, and do not address the confounding effects of changes in treatment; few have been validated in more than 1 cohort6–18. Moreover, there are no reports that have shown in longterm studies that changes in the level of these biomarkers reflect and anticipate changes in the risk for radiographic progression so that they can be considered valid surrogates to support their use both in clinical trials and to monitor patients in clinical practice19–28. A recent systematic review has highlighted the limitations of biomarker studies for prognosis in RA, especially the lack of standardization of tests for RA-related antibodies and differences in patient characteristics across studies evaluating specific biomarkers29.
The working group generated consensus on the following 5 objectives for an RA biomarker: (1) Change in the biomarker should reflect/predict change in the radiographic damage endpoint at the group level, so that the biomarker constitutes an endpoint for clinical trials and cohort studies, and at the individual patient level, so that the biomarker constitutes an endpoint for clinical practice. (2) Change in the biomarker should reflect/predict change in the damage endpoint independently of known predictors. (3) Change in the biomarker should correlate with the interval change in damage progression regardless of treatment approach. (4) The biomarker should be more responsive than routinely assessed clinical and laboratory measures associated with radiographic progression. (5) The biomarker should add prognostic information regarding radiographic progression over and above the combined information obtained from all other known predictors at both the group and individual patient level.
At OMERACT 9, this group used a Delphi approach to generate an international consensus for a minimum set of criteria regarding study design, principal outcomes, processing of biomarker samples, and documentation of potential confounders for the conduct of a prospective observational study with patients receiving therapeutic agents from different drug classes, aimed at the validation of a soluble biomarker reflecting damage endpoints according to these 5 objectives3,5. Such studies are essential so that a sufficiently broad spectrum of patients is included to verify external validity to the patient population typically seen in clinical practice as opposed to the highly selected patients recruited to randomized controlled trial. It was therefore considered essential in the study design to include patients with a wide spectrum of disease activity receiving diverse treatments but adhering to a treat-to-target (T2T) treatment strategy. This would serve not only to optimize patient outcomes but also to provide an opportunity to study the relationship between change in candidate biomarker(s) related to treatment and subsequent change in the radiographic endpoint.
The international RA BIODAM study is aimed at setting a benchmark for the design, implementation, and analysis of studies aimed at the validation of prognostic variables, including biomarkers, which are predictive of radiographic progression in RA. The data will also be used to derive risk assessment and prognostic tools for RA based on clinical and biological variables. In this report we provide details of the study design, baseline characteristics of patients recruited to the cohort, treatment received during the study, primary clinical outcomes, and radiographic progression over the 2 years of followup.
MATERIALS AND METHODS
Objectives of RA BIODAM
The primary objective of RA BIODAM was to generate data and study resources that would allow the conduct of an analysis to determine the independent predictive validity of soluble biomarkers considered to be high priority candidates for predicting structural damage in RA according to the criteria and protocol developed by the working group. It was agreed that secondary objectives would establish which clinical and laboratory predictors used in routine practice, individually and in combination, have the strongest and the most consistent association with change in radiographic damage. Tertiary objectives aimed to test the effect of treatment on biomarkers, to test statistical models to determine which may be optimal for describing the independent association between the biomarker and radiographic progression, and to establish sample size requirements for future studies of candidate biomarkers.
Study design of RA BIODAM
This was a multicenter, multinational, prospective observational study of patients with RA and fulfilling the 2010 Rheumatoid Arthritis Classification Criteria30. They were recruited consecutively from rheumatologist outpatient clinics and offices in Canada (n = 9), the United States (n = 5), Israel (n = 1), Denmark (n = 1), France (n = 6), Germany (n = 4), Ireland (n = 1), Italy (n = 6), the Netherlands (n = 4), and Norway (n = 1); trial registration: Assess Structural Damage in Rheumatoid Arthritis Using Biomarkers and Radiography: Clinicaltrials.gov: NCT01476956, clinicaltrials.gov/ct2/show/NCT01476956. The first patient was recruited on October 30, 2011, and the last patient visit was May 17, 2017. Principal study design features were focused on recording change in biomarker following change/institution of conventional synthetic disease-modifying antirheumatic drugs (csDMARD) therapy [methotrexate (MTX), sulfasalazine, hydroxychloroquine, chloroquine, leflunomide] and/or following addition of tumor necrosis factor inhibitor (TNFi) therapy [adalimumab (ADA), etanercept, infliximab (IFX), certolizumab pegol, golimumab] as patients were observed every 3 months over a 2-year time frame. Consequently, the study recruited patients who were (1) starting csDMARD therapy; or (2) changing csDMARD therapy (defined as an increase in dose of MTX by ≥ 5 mg weekly to a maximum dose of 25 mg weekly, add-on of an alternative csDMARD, switch to an alternate csDMARD); or (3) starting TNFi therapy alone or in combination with csDMARD therapy.
Disease activity was monitored systematically every 3 months using the 44-joint count Disease Activity Score (DAS44). Changes in csDMARD and/or TNFi therapy were to be implemented according to the 2010 European League Against Rheumatism recommendations, which call for a target of remission (DAS44 < 1.6) for patients receiving csDMARD therapy in the setting of early disease (< 2 yrs disease duration) and a target of low disease activity state (DAS44 ≤ 2.4) for patients receiving TNFi in the setting of established disease and prior exposure to csDMARD31. Biosamples were collected every 3 months and prior to a change in csDMARD and/or TNFi therapy. Change in TNFi therapy was prespecified as an increased dose of IFX (3–5 mg/kg) and/or frequency (every 8 to every 6 weeks), an increased frequency of ADA (every other week to weekly), a switch to a different TNFi, or a switch to an agent from a different class of biological DMARD (bDMARD; rituximab, abatacept, tocilizumab).
High-dose oral glucocorticoid therapy (as in the COBRA trial32) could be implemented in early RA at the start of treatment with serum/urine biomarker samples being obtained prior to implementation and on a monthly basis until therapy had been stable for at least 1 month at ≤ 10 mg prednisone daily. Intraarticular steroid injections could be administered as required with a biosample being obtained prior to and 1 month after administration if the total dose was ≥ 40 mg methylprednisolone (or equivalent).
The study fulfilled Good Clinical Practice Guidelines, complied with the Declaration of Helsinki, and received ethical approval from the local ethics committees of each of the 38 sites (Supplementary Table 1, available from the authors on request). All patients provided written informed consent.
Inclusion and exclusion criteria
Consecutive patients with RA fulfilling the 2010 RA Classification Criteria were recruited30. Inclusion criteria were age of 18 years or older, joint symptoms for at least 3 months, DAS44 ≥ 2.4, and about to start or change csDMARD therapy or to start TNFi therapy. If already receiving csDMARD therapy, this had to be stable for 1 month prior to baseline, and if on systemic steroid (prednisone ≤ 10 mg/day allowed), this had to be stable for 1 month prior to baseline. Patients were excluded if they had already received treatment with a TNFi or other bDMARD. Additional exclusion criteria are listed in the Supplementary Data, available from the authors on request.
Data collection
The following assessments were conducted at screening/baseline: age, sex, marital status, education, smoking history, ethnicity, alcohol use, recreational drug use, age at disease onset, symptom duration, number of criteria met for classification of RA, current and past medical history, past RA medication history, current treatment with csDMARD and/or steroids (dose, frequency, start date), approximate duration of treatment with current and previous csDMARD and steroids, 44 swollen and 53 tender joint counts, DAS44 based on erythrocyte sedimentation rate (DAS44-ESR), physician global numerical rating scale (NRS; 0–10), patient self-report questionnaires [pain NRS (0–10), patient global NRS (0–10), Health Assessment Questionnaire (HAQ), fatigue NRS (0–10), Medical Outcomes Study Short Form-36, RA Impact of Disease Score33, RA Flare Questionnaire], Revised American College of Rheumatology (ACR) Functional Classification of Global Functional Status in RA34, height, weight, vital signs, ESR, C-reactive protein (CRP), rheumatoid factor (RF; IgM), anticitrullinated protein antibodies (ACPA; assessed by anticyclic citrullinated peptide antibody assay), HLA-DRB1 genotype, radiographs of hands and feet, chest radiograph, and tuberculosis testing for patients about to start TNFi therapy. Efficacy outcomes were also conducted at followup visits every 3 months and early termination visits as well as current RA treatment (dose, frequency, start/stop date, reason for discontinuation) and treatment-emergent adverse events (Medical Dictionary for Regulatory Activities coding system). Greyscale and power Doppler ultrasonography (US) of hands and feet were conducted on a subset of patients every 3 months using the German US 7 Score35.
Radiographic assessment
Radiographs of hands (postero-anterior) and feet (antero-posterior) were obtained every 6 months using a standardized methodology and submitted centrally. Radiographic joint damage was assessed centrally according to the Sharp/van der Heijde (SvdH) method (range 0–448)36 by 2 trained assessors independently, who were blinded to the patient’s identity, treatment, and treatment center, but who were aware of the chronology of the films with the mean score of the 2 assessors being used for analyses of the primary endpoint. Progression of radiographic joint damage over 6-month, 12-month, and 2-year intervals was prespecified as a change in radiographic score greater than the smallest detectable change (SDC), as well as by a change (in the total radiographic score) > 0.5. Adjudication of discrepant radiographs by a third reader was prespecified on the basis of a 1-year change score and SDC. This was conducted if the mean 1-year change score was > SDC but change score for 1 of the readers was < SDC. The final scores for status and change scores for discrepant cases were then derived by calculating the average of the adjudicator scores and the scores of the primary reader with the closest value for the 1-year change score.
Biosample collection, transportation, processing, and storage
Serum/urine biomarkers were obtained every 3 months. A biosample manual specified procedures to ensure standardized acquisition and processing of biosamples across all sites. A blood sample for serum was taken 2–4 h after rising and processed according to the OMERACT recommendations for the minimal handling of biomarker samples3. RNA samples were acquired using PAXgene tubes in all patients at Canadian study sites, and at other sites prior to the first TNFi dose and 1 month later. Biosamples were stored locally at −70°C without any further processing until batch-shipped at −150°C in liquid nitrogen vessels using Cryoport Inc., which provides sample temperature monitoring throughout the entire shipping process. The biosamples were shipped to the RA BIODAM biorepository at the CaRE Arthritis coordinating site (Edmonton, Canada) where they were thawed, processed into 0.5 ml aliquots, and stored at −70°C.
Study endpoints
The primary endpoint of the RA BIODAM study was radiographic joint progression according to the SvdH method (range 0–448). Secondary endpoints include the SvdH erosion score (range 0–280), and the 3- and 6-month change in biomarker level from baseline following the introduction of csDMARD or TNFi therapy.
Sample size
The sample size calculation was based on detecting a relationship between a specified biomarker and the target outcome, and used the approach to sample size calculation for logistic regression of Hsieh, et al37. A survey was conducted among the OMERACT RA BIODAM working group to obtain estimates for 4 variables needed for the calculation: probability of no progression given that the biomarker is not normal, odds of no progression when the biomarker is normal to odds of no progression when the biomarker is not normal, proportion of the sample (or population) with no progression, and the amount of variation of the biomarker that is explained by the other covariates in the model (SvdH, DAS44, age, sex, ACPA, HLA-DRB1-SE, and interactions). A logistic regression of a binary target variable on a binary biomarker variable with a sample size of 600 observations (of which 50% are expected to not progress given the biomarker is not normal) would achieve 90% power at a significance of 0.05 to detect a biomarker with an OR > 3 for no progression when biomarker is normal to odds of no progression when biomarker is not normal. An adjustment for the amount of variation of the biomarker that is explained by the other covariates in the logistic regression was needed for the sample size calculation; this was estimated to be an R2 of 0.25 based on the survey results.
Statistical analysis
We used descriptive data to report baseline cohort demographics and disease characteristics, clinical outcomes (DAS44, HAQ) and percentage achieving clinical remission (DAS44, ACR Boolean38) over 2 years, and cumulative probability plots for radiographic progression. Treatment categories over the course of followup comprised the following patient groups: (A) started taking csDMARD and continued csDMARD; (B) started taking csDMARD and switched to TNFi; (C) taking csDMARD at baseline and continued csDMARD; (D) started csDMARD at baseline and switched to TNFi; (E) started TNFi and continued first TNFi; (F) started TNFi and switched to another TNFi; and (G) started TNFi and switched to non-TNFi bDMARD. We combined data from patient categories A and C into 1 group (csDMARD-treated only); B and D into a second group (csDMARD switched to TNFi); and E, F, and G into a third group (started on TNFi and continued with bDMARD treatment). Missing data was imputed using the last observation carried forward for continuous outcomes and nonresponder imputation for dichotomous outcomes. Statistical comparisons of treatment groups were not conducted because this was not prespecified and was not considered among the objectives of the study.
RESULTS
Baseline demographics and disease status
Complete baseline data was available on 571 patients who were recruited from Oct. 30, 2011, and last patient visit was on May 17, 2017. Each center (n = 38) recruited between 1 and 60 patients (median 10.5) and 439 (76.9%) had complete 2-year followup. Reasons for discontinuation were subject withdrew consent (52), subject lost to followup (25), major protocol violation(s) with study noncompliance (14), subject noncompliant with protocol (13), serious adverse event (10), other/unspecified (10), worsening of intercurrent medical condition (5), and investigator judgment (3). Baseline patient and disease characteristics comparing completers and noncompleters are shown in Table 1. Overall, the patient population comprised a demographically typical cohort of patients with RA, the majority being female (76%) and with mean age of 55.7 years. Mean disease duration was 6.5 years and 52% had prior exposure to a csDMARD. Patients had active disease at baseline with a mean of 8.4 swollen joints, 13.6 tender joints, DAS28 of 5.2, and DAS44 of 3.8. The majority (77.7%) were either RF- or ACPA-positive with a mean CRP of 14.9 mg/l at baseline. Patients with complete 2-year followup had fewer comorbidities, lower levels of disease activity (DAS44, Simplified Disease Activity Index, swollen joint count, ESR), and were more likely to be treated with oral steroids at baseline.
Baseline patient and disease characteristics comparing completers and noncompleters in the RA BIODAM Cohort.
Serum, urine, and radiographs were obtained from 4638 (90.3% complete), 4591 (89.3% complete), and 2343 (82.1% complete) visits, respectively. US scores were obtained from 1034 visits and 130 patients had at least 1 assessment at 11 study sites.
The percentage of patients treated with csDMARD remained stable over time at about 90% while the number taking bDMARD increased from 41% to 52% (Supplementary Figure 1, available from the authors on request). There were 142 patients (24.9%) who were naive to csDMARD at baseline who then started taking csDMARD, and 30 (5.3%) were additionally started on TNFi therapy during the followup. Of 195 patients (34.2%) already taking csDMARD therapy at baseline, 40 (7.0%) were additionally started on TNFi therapy during followup. There were 231 (40.5%) patients who began TNFi at baseline, 195 of which were taking concomitant csDMARD therapy. Of these, 36 switched to an alternate TNFi, and 26 switched to a non-TNFi bDMARD. The percentage of patients taking oral steroids decreased from 45% at baseline to 26% at followup.
DAS and HAQ scores decreased in the first 6 months of treatment, although this was less evident in those patients taking csDMARD who subsequently received TNFi therapy (Figure 1 and Figure 2). The percentage of patients achieving DAS and ACR Boolean remission gradually increased over the course of 2 years, this again being less apparent in those taking csDMARD who subsequently received TNFi therapy (Figure 3 and Figure 4).

DAS44 over 2 years in the RA BIODAM cohort according to treatment category. A. Missing data imputed by LOCF. B. Observed data. DAS44: 44-joint count Disease Activity Score; RA: rheumatoid arthritis; LOCF: last observation carried forward; csDMARD: conventional synthetic disease-modifying antirheumatic drug; TNFi: tumor necrosis factor inhibitor.
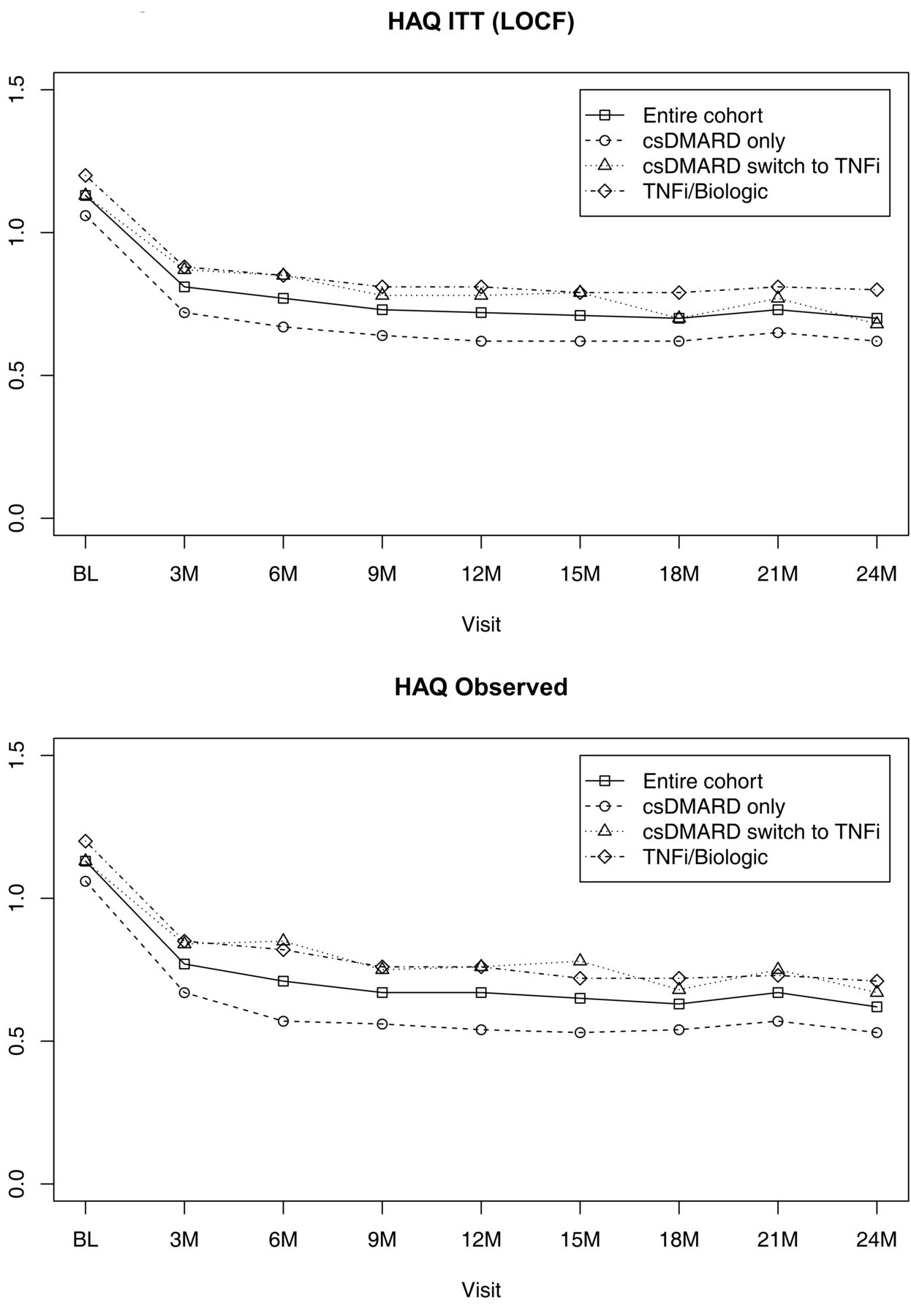
HAQ over 2 years in the RA BIODAM cohort according to treatment category. A. Missing data imputed by LOCF. B. Observed data. HAQ: Health Assessment Questionnaire; RA: rheumatoid arthritis; ITT: intent-to-treat; LOCF: last observation carried forward; csDMARD: conventional synthetic disease-modifying antirheumatic drug; TNFi: tumor necrosis factor inhibitor.

Percentage of patients achieving DAS44 remission over 2 years in the RA BIODAM cohort according to treatment category. A. NRI analysis. B. Observed data. DAS44: 44-joint count Disease Activity Score; RA: rheumatoid arthritis; NRI: nonresponder imputation; csDMARD: conventional synthetic disease-modifying antirheumatic drug; TNFi: tumor necrosis factor inhibitor.

Percentage of patients achieving ACR Boolean remission over 2 years in the RA BIODAM cohort according to treatment category. A. NRI analysis. B. Observed data. ACR: American College of Rheumatology; RA: rheumatoid arthritis; NRI: nonresponder imputation; csDMARD: conventional synthetic disease-modifying antirheumatic drug; TNFi: tumor necrosis factor inhibitor.
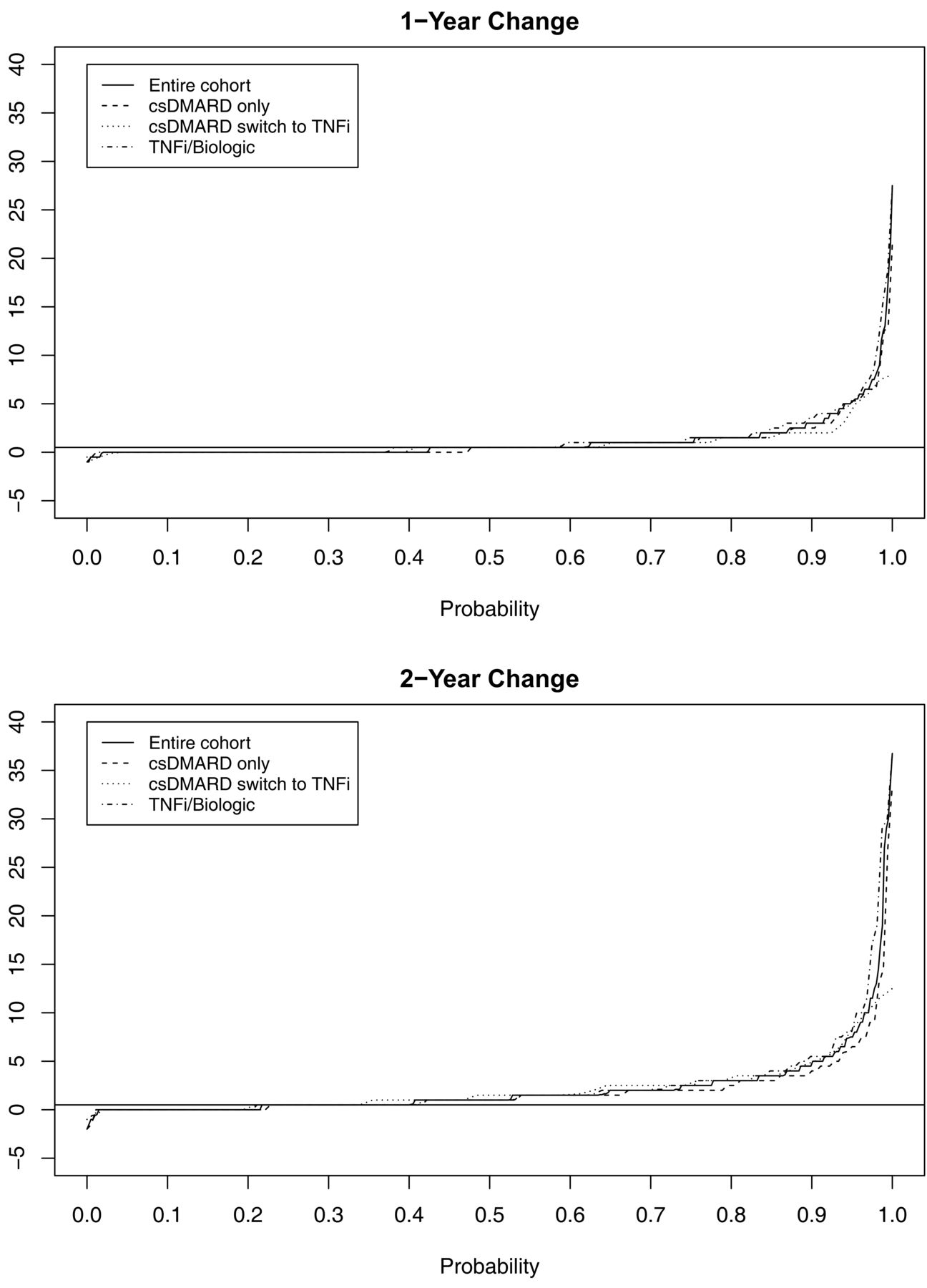
Radiographs from baseline and 1 year were available in 442 patients and from baseline and 2 years in 406 patients. Mean SvdH score at baseline, 1 year, and 2 years was 17.2, 18.5, and 20.0, respectively for those completing 1- and 2-year followup. SDC for radiographic progression was calculated to be 2.6 SvdH units and progression > 2.6 units was observed in 48 (10.9%) at 1 year and in 91 (22.4%) at 2 years. Radiographic progression of > 0, > 0.5, ≥ 3, and ≥ 5 SvdH units was observed in 255 (57.8%), 169 (38.2%), 48 (10.9%), and 27 (6.1%) at 1 year, and in 318 (78.3%), 243 (59.9%), 91 (22.4%), and 41 (10.1%) at 2 years of followup, respectively (Figure 5A and 5B). There were few differences between the categories of treatment.

Cumulative probability plot of radiographic progression assessed with the SvdH according to treatment category. A. 1-year progression. B. 2-year progression. SvdH: Sharp/van der Heijde score; csDMARD: conventional synthetic disease-modifying antirheumatic drug; TNFi: tumor necrosis factor inhibitor.
DISCUSSION
This is the first report of the data from RA BIODAM, which was an investigator-initiated 6-year international collaborative effort to compile a unique resource of clinical and imaging data with biosamples acquired according to an international consensus for the conduct of a prognostic study and standard operating procedures for the handling, transportation, and storage of biosamples. In particular, the procedures developed for RA BIODAM ensured that biosamples from 90% of patients from 10 countries were maintained at −70°C from the point of blood draw and serum extraction to the point of creating aliquots for storage in the central biorepository in Canada. Moreover, the level of patient retention, data collection, and acquisition of radiographs over 2 years was at least comparable to many clinical trials that included on-site study oversight and source data verification39,40.
Our data demonstrate that the RA BIODAM Cohort is characteristic of patients with RA starting DMARD therapy in current clinical practice regarding both demographics and disease status and is therefore an appropriate cohort for the validation of biomarkers. Disease activity and severity was somewhat lower than observed at baseline in recent clinical trial cohorts, although patient selection for RA BIODAM only required DAS > 2.4 as a disease activity variable. A majority of patients were female, and symptom duration was about 6 years. Disease was active with an average of 8 swollen and 13 tender joints and DAS44-ESR of almost 4, and almost 80% were serologically positive for either RF or ACPA. Just over half had already been exposed to csDMARD and about 40% began TNFi agents at baseline.
A limitation of the study is that patients who withdrew had more swollen joints and higher acute-phase reactants, but fewer used corticosteroids. With regard to bias relative to the objectives of this study, it is possible that these patients reflect a relatively refractory cohort of patients with inadequate responses to treatment and more likely to demonstrate radiographic progression. This could limit detection of an association between certain types of biomarkers reflecting inflammation and progression of radiographic damage. However, progression > 0.5 was evident in 60% of patients at the 2-year followup. Another limitation is that the loss of these patients precludes an opportunity to examine physician- and patient-related factors associated with failure to adhere to the treatment strategy. However, physicians had considerable discretion regarding the type of therapy that could be instituted should patients fail to achieve the DAS target. Clinical responses and radiographic progression were comparable to those observed in previously reported cohorts that used a T2T strategy41–47.
The RA BIODAM investigators have completed a 2-year prospective study that recruited patients with characteristic demographic and disease features of RA, culminating in an extensive list of clinical, imaging, and biosample resources that will permit the clinical validation of candidate biomarkers for radiographic damage endpoints. The resources generated in RA BIODAM will be made available to the research community to help expedite the identification and validation of such biomarkers.
Acknowledgment
The authors acknowledge the contribution of the following RA BIODAM investigators and their local site teams: Thomas Neumann, Department of Internal Medicine III/Rheumatology/Osteology, Universitätsklinikum der Friedrich-Schiller-Universität Jena, Jena, Germany; Christopher Ritchlin, Division of Allergy/Immunology & Rheumatology, University of Rochester Medical Center, Rochester, New York, USA; Joan Bathon, Division of Rheumatology, Columbia University Medical Center and Columbia University College of Physicians and Surgeons, New York, New York, USA; René-Marc Flipo, Centre d’Investigation Clinique, CHRU de Lille, Lille, France; Proton Rahman, Memorial University, St. John’s, Newfoundland and Labrador, Canada; Carol Hitchon, Arthritis Center, Department of Internal Medicine, Rheumatology Section, University of Manitoba, Winnipeg, Manitoba, Canada; Saeed Shaikh, Niagara Peninsula Arthritis Centre Inc., St. Catharines, Ontario, Canada; Wolfgang Spieler, Osteology and Rheumatology, ZeFOR GmbH Center for Research, Zerbst, Germany; Leonardo Punzi, Department of Medicine, Università di Padova, Padua, Italy; Ingo Tarner, Department of Rheumatology and Clinical Immunology, Kerckhoff-Klinik, Bad Nauheim, Germany; Vivian Bykerk, Inflammatory Arthritis Center, Hospital for Special Surgery, New York, New York, USA; Philip J. Mease, Seattle Rheumatology Associates, Seattle, Washington, USA; Renée Allaart, Department of Rheumatology, Leiden University Medical Centre, Leiden, the Netherlands.
Footnotes
This is an investigator-initiated study aimed at the clinical validation of biomarkers, which has been supported by unrestricted funding from AbbVie Corp. AbbVie had no role in the design, execution, or analysis of this study and had no role in the drafting of the manuscript.
- Accepted for publication August 12, 2019.
REFERENCES
DATA SHARING POLICY
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Three different databases were developed by the coordinating project management group CaRE Arthritis Ltd., which were linked by the patient study ID:
Clinical database: clinical data was recorded in the RA BIODAM electronic case report form, and an interactive system of study queries was used to proactively verify data entry and address missing data within prespecified time frames.
Biosample biorepository: aliquoted sera, urine, and RNA biosamples were barcoded and stored at −70°C.
Imaging repository: all anonymized Digital Imaging and Communications in Medicine (DICOM) radiographs of hands and feet passed quality assurance procedures.
Access to all RA BIODAM data and biosamples will be made available for academic and not-for-profit entities. This will require the submission of a study proposal to the scientific committee; see www.carearthritis.com.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}