

Abstract
Objective. Clinical data suggest that infections can trigger IgA vasculitis (IgAV), but longterm observations are lacking. We compared rates, types, and microorganisms for serious infection before and after diagnosis for children with IgAV and non-exposed controls.
Methods. Using population-based administrative linked health datasets we estimated incidence rates (IR) for serious infection per 1000 person-months for patients with IgAV (n = 504, age 5 yrs, 59.1% males) and controls matched for age, sex, and year of presentation (n = 1281, age 6 yrs, 66% males). Time zero (T0) was the date of IgAV diagnosis or equivalent date in controls, lookback (median 38 mos) was the period prior to T0, and followup (median 239 mos) was the period after T0.
Results. During lookback, prevalence of serious infection was similar in patients with IgAV and controls (11.5% vs 9.5%, respectively), but patients with IgAV had a higher rate of upper respiratory tract infections [incidence rate ratio (IRR) 1.79; 95% CI 1.39–2.31] with shorter time between first serious infection and T0 (27 vs 43 mos; p = 0.02). During followup, patients were at a constant increased risk for serious infections (IRR 1.46, 95% CI 1.35–1.58). These rates were higher during followup: sepsis (IRR 12.6), pneumonia (IRR 6.19), upper respiratory tract infections (IRR 2.36), and skin infections (IRR 1.85). There was little overlap between patients with serious infections in the lookback and followup periods.
Conclusion. In patients with childhood IgAV there is an increased longterm risk for a broader spectrum of infections, which is unrelated to serious infections prior to diagnosis or treatment. This suggests disease-specific factors may have a lasting effect on immune competence in childhood IgAV.
IgA vasculitis (IgAV), formerly known as Henoch–Schönlein purpura, is a multisystem vasculitis that occurs most frequently in children and typically presents with purpuric skin lesions due to IgA-induced leukocytoclastic vasculitis, but can also lead to arthritis, enteritis, and glomerulonephritis1,2. Clinical observations suggest a temporal relationship between IgAV and infections, because the parents of 30%–50% of patients with IgAV give a history of recent upper respiratory tract infection (URTI) and the incidence of IgAV increases during colder seasons3,4,5. However, while URTI are very common in children, only a minority develop IgAV. Further, because IgAV case series have reported a broad spectrum of potential infectious triggers, no clear relationship exists between a specific microorganism and IgAV development6. This suggests that in addition to an infectious trigger, other factors are needed for disease development. Diminished glycosylation of mucosal polymeric IgA caused by genetic anomalies or induced by certain bacteria has emerged as an important factor in the pathogenesis of IgAV7,8,9,10,11,12,13,14, while previous studies suggested a role for genetic factors, such as carrying HLA class I (B*41:02) and class II (DRB1*01), and/or changes in Toll-like receptor 9 or cytokines15,16,17,18,19. Genetic anomalies are likely to be persistent and would leave affected patients with IgAV susceptible to more infections over time10,16.
Based on these considerations, we investigated the variation over time in the rate and type of infections leading to hospital presentation for patients with childhood IgAV and matched controls.
MATERIALS AND METHODS
Study cohort
Data were obtained from the Hospital Morbidity Data Collection and the Emergency Department Data Collection, which contain routinely collected data for all hospitalizations and emergency department visits in the state of Western Australia. These datasets are effectively linked through a combination of probabilistic matching and clerical review20,21. This population-level observational study included persons < 17 years of age with a hospital admission or emergency department presentation for IgAV [International Classification of Diseases (ICD)-9-CM 278.0; ICD-10-AM D69.0], who lived in Western Australia between January 1, 1980, and December 31, 2015. For each patient with IgAV, we included up to 3 non-exposed controls free of rheumatic disease conditions and matched for year and semiannual month of birth, sex, and indigenous status. Extraction of all available data resulted in a dataset with information on demographics, principal and up to 21 secondary diagnoses, and principal and up 10 secondary procedures, length of stay, and emergency department discharge codes for the total study sample. Because the first hospital contact was not necessarily for IgAV, we defined a time zero (T0), which for patients with IgAV (n = 503) was the first date with an IgAV diagnostic code, and for each control (n = 1281) the date that most closely mirrored the index (diagnosis) timepoint for the matched patient with IgAV. A lookback period was defined as all observation time prior to T0, while a followup period was defined as all observation time following T0. Serious infections were defined as episodes leading to emergency department presentation and/or hospital admission resulting in an infectious disease code. Given the wide scope of possible infections and to allow comparison with previous studies, we included 4 of 5 infectious categories (pneumonia, sepsis or bacteremia, urinary tract infection, and skin and soft tissue infections) proposed by Tektonidou, et al22; but we excluded opportunistic infections because they were deemed less relevant for this age category (Supplementary Table 1, available from the authors on request). Further, we analyzed the presence of a number of specific infections including laryngitis or pharyngitis, upper respiratory tract infections (URTI), and microorganisms in accord with the study by Weiss, et al23 (Supplementary Table 2, available from the authors on request) and the presence of rarer infectious agents that have been proposed as triggers for IgAV6 (Supplementary Table 3).
This study was approved by the Human Research Ethics Committee at the Western Australia Department of Health (HREC 2016.24).
Statistical analysis
Descriptive statistics include median and interquartile range (IQR) for continuous variables, which were compared by nonparametric methods (Kruskal-Wallis test); and categorical data are described as a frequency and proportion, and compared with an OR with a 95% CI or by chi-square with Yates correction when needed. Infection incidence rates (IR) were calculated per 1000 person-months with infection IR ratios (IRR) derived from conditional maximum likelihood estimates using Byar’s method for Poisson distribution24. To determine which of 8 clinical characteristics [sex, age, indigenous background, presence of renal or gastrointestinal indications, increased erythrocyte sedimentation rate (ESR) at diagnosis] could be potential independent risk factors for the occurrence of serious infections in the followup period in the IgAV cohort, we used backward logistic regression (p < 0.1 to enter, p < 0.05 to stay). All statistics were derived from SPSS v23.0 and OpenEpi software, with 2-sided p values < 0.05 considered statistically significant.
RESULTS
There was comparable male preponderance and distribution of rural versus metropolitan hospital contacts in both cohorts. Controls were slightly older at T0 (6 vs 5 years of age; p < 0.01), while patients with IgAV more often had joint, skin, and renal symptoms, and a longer length of stay, and there was a higher proportion of indigenous Australians. Demographic and clinical data for patients with IgAV and non-exposed controls at T0 are given in Table 1.
Demographic and clinical data for patients with IgA vasculitis (IgAV) and non-exposed controls at time zero.
During the lookback period, hospital contacts were logged for 314/504 (62.3%) patients with IgAV and 982/1281 (76.7%) controls. The proportion of patients with IgAV and controls with any serious infection was equivalent (11.5% vs 9.7%, respectively; p = 0.32; Table 2), but the observation-time adjusted rate of a serious infection was greater in patients with IgAV than in controls (IRR 1.37; 95% CI 1.06–1.77). This was driven by throat infections (IRR 16.07; 95% CI 2.25–382.0) and URTI (IRR 1.79; 95% CI 1.39–2.31), which occurred at significantly higher rates in patients with IgAV, while other infection types did not (Table 2). The median time from first serious infection to T0 was shorter in patients with IgAV (27 mos; IQR 9–70) than in controls (43 months; IQR 21–77; p = 0.022).
Observed incidence rates (IR) and IR ratio (IRR with 95% CI) overall and by primary infection site in the lookback period in patients with immunoglobulin A vasculitis (IgAV) and controls.
A total of 20 patients with IgAV (4.1%) and 5 controls (0.4%; p < 0.01) had a serious infection concurrent with diagnosis/T0, and these serious infections were excluded from the followup data analysis, as were 12 patients with IgAV (2.3%) and 45 controls (3.5%) with no further hospital contacts following T0 (p = 0.22). During a mean followup of 256 months (IQR 189–324), 141/492 (28.7%) of the remaining patients with IgAV and 241/1236 (21.1%) of the controls developed a new serious infection (OR 1.50, 95% CI 1.18–1.90; Table 3), with only a minority of patients with IgAV and controls (14.2% vs 13.1%, respectively; p = 0.3) having a serious infection in both the lookback and the followup period. Excluding the 2 patients with IgAV (0.3%) who were receiving chronic dialysis reduced this only marginally (OR 1.39, 95% CI 1.06–1.83). In logistic regression analysis, male sex (OR 1.55, 95% CI 1.21–1.99, p < 0.001) and diagnosis of IgAV (OR 1.32, 95% CI 1.02–1.69, p = 0.035) were independently associated with occurrence of serious infection during followup, with no effect detected for age at T0, rural residence, indigenous status, or the presence of renal, joint or gastrointestinal involvement at T0 (all p > 0.1; data not shown). When restricting this analysis to patients with IgAV, male sex (OR 1.55, 95% CI 1.03–2.31, p = 0.035) and increased ESR at T0 (OR 2.09, 95% CI 0.99–4.40, p = 0.051) emerged as independent predictors for serious infection during followup.
Observed incidence rates (IR) and IR ratio (IRR with 95% CI) overall and by primary infection site after time zero (T0) in patients with immunoglobulin A vasculitis (IgAV) and controls.
With a total of 639 serious infection events, the infection rate per 1000 months of followup for patients with IgAV was 2.51 (2.22–2.83) versus 1.13 (1.01–1.25) for controls, for a serious infection RR of 1.46 (1.35–1.58). Both the time to a first serious infection (Figure 1; p = 0.29) and the accrual of serious infections over time were similar for patients with IgAV and controls (Figure 2; p = 0.3). Multiple serious infections occurred in 69/141 (48.9%) patients with IgAV and 66/241 (27.3%) controls (p < 0.01). In the followup period, patients with IgAV had greater likelihood of sepsis (IRR 12.6), pneumonia (IRR 6.19), throat infections (IRR 2.47), URTI (IRR 2.36), and skin infections (IRR 1.85). Further, the rates for all types of infections during followup were higher than in the lookback period, except for URTI (Table 3). While the absolute number of infections in which a microorganism was identified was low (Table 4), the likelihood of confirmed Group A streptococcal infection was higher in patients with IgAV before diagnosis (IRR 16.07, 95% CI 2.23–382.9), while during followup patients with IgAV were more likely to incur Haemophilus influenzae (IRR 15.7), mycoplasma (IRR 6.3), parainfluenza (IRR 3.47), staphylococcal (IRR 2.7), or salmonella (IRR 6.3) infections.
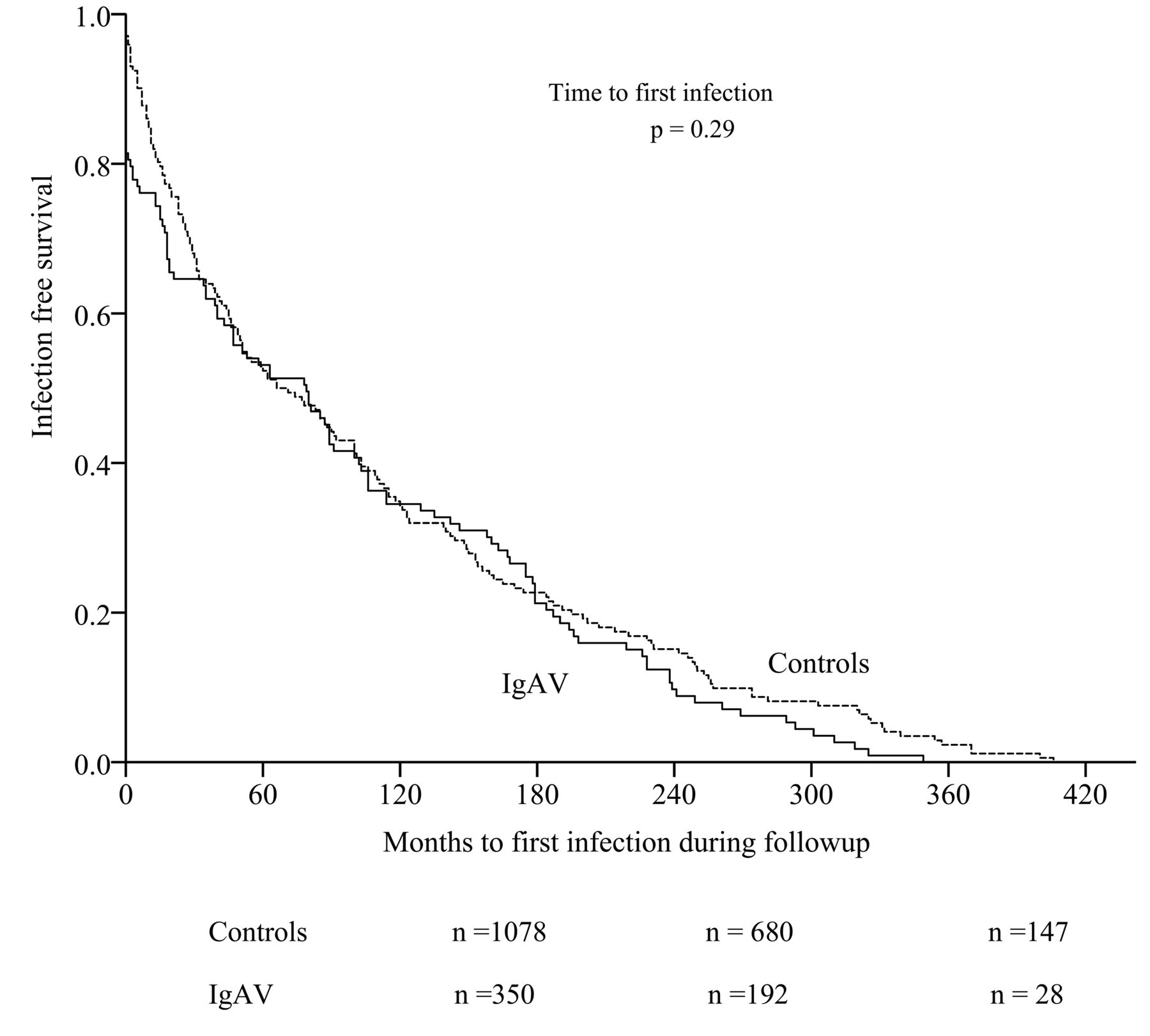
Survival curves show time to a first infection after diagnosis/T0 in patients with IgAV (n = 492) and controls (n = 1236) during the followup period. Numbers under the graph indicate no. patients remaining in the study at that time. T0: time zero; IgAV: IgA vasculitis.

Accrual over time of serious infections (SI) in patients with IgAV (SI = 265) and controls (SI = 374) during the followup period. Numbers along Y axis are in hundreds. IgAV: IgA vasculitis.
Incidence rates and incidence rate ratio (with 95% CI) by microorganism isolated for lookback and followup periods in patients with IgA vasculitis (IgAV) and controls.
DISCUSSION
In this population-level study, the proportion of childhood patients with IgAV presenting with any serious infection prior to diagnosis was equivalent to a matched control population. However, throat infections and URTI occurred at a higher time-adjusted rate and closer to the time of diagnosis in patients with IgAV, with Group A streptococcus as the main microorganism. During longterm followup after diagnosis, patients with IgAV were at increased risk of serious infections, with greater incidence rate and diversity of primary infection sites and microorganisms observed than before diagnosis.
The study cohort included all patients with IgAV presenting to a metropolitan or rural healthcare facility in Western Australia over the study period. The age and sex distribution as well as the predominant presentation during colder months for this IgAV cohort is in agreement with other case series, and indicates that our administrative dataset identified a representative cohort of childhood patients with IgAV3,25,26. While the cause of IgAV remains largely unexplained, case series indicate that a history of URTI during the preceding one to 2 weeks can be elicited in 30% to 70% of those with childhood IgAV, but confirmation of infection (by culture or serology) is obtained in fewer than half of these cases4,23,27,28. Our data indicate that serious infections resulting in hospital presentation occur in 11.5% of cases prior to diagnosis during an extended period of time of up to 2 years prior to development of IgAV. Combined with the higher incidence rate of a Group A streptococcal-related throat infection and URTI, this suggests that recurrent specific infections may set off abnormal IgA production long before the emergence of clinical disease. This is reminiscent of the progression from innocuous antibody formation to development of seropositive rheumatoid arthritis29. While overproduction of IgAV has been related to innate predisposing factors such as Toll-like receptor and cytokine polymorphisms15,16,17,18,30,31, the similar proportion of patients with IgAV and controls presenting a serious infection before T0 argues against a predisposing role for such potential underlying immune abnormalities, especially in circumstances in which Group A streptococcus serves as a repeated trigger for an IgA response. A recent large Korean study indirectly associated IgAV presentation with the seasonal occurrence of viral infections5; however, with the low numbers of confirmed infection, we found no role of viral inducers as triggers for IgAV. Rates for other infection types preceding IgAV were low, overall, with causative microorganisms as infrequently identified in this and other studies3,23,28, but our data nonetheless revealed similarly low rates in patients with IgAV and controls.
IgAV in children is considered a benign, self-limiting disease often leading to complete remission32. Nonetheless, to reduce troublesome symptoms and prevent organ damage, between 25% and 70% of children with IgAV are temporarily treated with corticosteroids and/or immunosuppressive drugs33,34,35. While this confounds the relationship between infections and IgAV, particularly in the first year after diagnosis, most patients will be taken off these medications after 1 year36. In spite of this, we demonstrate that patients with IgAV remain at higher risk for further infections for at least 20 years post-diagnosis (Figure 1). In addition, the spectrum of primary infection sites becomes much broader following IgAV diagnosis (Table 3), together with broader diversity in the causative microorganisms (Table 4). While comparative studies are not available, it seems unlikely that this longterm increased risk of infection is a direct effect of temporary immune-modulating treatment. Together with the marginal overlap between patients with serious infections before and after diagnosis, this suggests that overall immune competence in patients with IgAV may become compromised around the time of diagnosis. Whether this relates to mucosal persistence of abnormally glycosylated IgA and/or to increased expression of immune response proteins, to other acquired deficiencies, or to a combination of these is speculative. In addition to development of IgAV, we found that the odds of serious infection during followup were increased by 50% for male patients, and by 100% when ESR was elevated at diagnosis of IgAV. These findings are somewhat surprising because no effect of other baseline disease manifestations was seen, but they are in agreement with studies describing an overrepresentation of males among young patients hospitalized with URTI37,38. The increased odds of serious infections with elevated baseline ESR could reflect the burden of inflammation at T0, but because ESR is usually slow to rise in the acute phase, the increased ESR could indicate a longer disease duration. One could speculate whether the increased baseline ESR in IgAV indicates a preceding buildup of excessive IgAV levels as part of the abnormal immune response, rather than a response to the infection, which mirrors the experience of patients with chronic arthritis39,40,41.
Our results should be considered in light of methodological limitations. While the use of administrative data ensured state-wide identification of patients with IgAV, it relies on the accuracy and validity of coding, which has some limitations and does not allow a detailed analysis of clinical, microbial, or serological findings. Also, this was an observational study, with no uniform approach to the diagnosis and management of IgAV and associated infections, and no data on vaccination rates and completeness. Difficulties in selecting sufficient controls from electoral rolls for this very young cohort led to a somewhat flawed matching process, with a mild imbalance regarding age at T0 and indigenous status. With small subgroup numbers, our study may have suboptimal power where type I errors cannot be excluded. Importantly, controls were not healthy individuals, but represented individuals seeking hospital care for a range of conditions other than rheumatic diseases. This may have inflated the number of controls with infections and led to underestimation of the infection risk for IgAV, in contrast to use of a true healthy cohort. Also, our study does not address the question of what triggers IgAV immune complex formation in patients without an obvious recent infection. Also, while we selected a limited number of clinically relevant independent variables and there was no collinearity, logistic regression modeling requires a uniform and constant relationship over the entire range of values between the predictor and the dependent, and this assumption does not hold true for the low age range in our 2 cohorts. Further, selection of variables in explanatory modeling can compromise the stability of a final model. Because there is no appropriate correction for the repeated model fitting, stepwise logistic regression modeling may lead to p values that are too low owing to multiple comparisons and CI that are too narrow around the parameter estimates. Finally, our selection of patients with IgAV excluded patients managed in general practice, and this may have biased our findings because these patients likely have a less severe disease presentation.
The strengths of our study lie in the population-based approach, with a large sample size, the inclusion of a matched control group, and the longterm observations both before and after diagnosis.
URTI and throat infections were the only infections occurring at higher rates in patients with IgAV in a 2-year period before diagnosis. Male patients with IgAV remained at increased risk for a wide range of infections for up to 20 years after IgAV diagnosis, with no obvious carryover effect of earlier infection or immune-modulating treatment. This suggests lasting changes in immune competence around the time of diagnosis of IgAV.
Acknowledgment
The authors thank the data custodians of Hospital Morbidity Data Collection and Emergency Department Data Collection and staff at the Western Australian Data Linkage Branch for their assistance in provision of data.
Footnotes
Supported by an unrestricted grant from the Arthritis Foundation of Western Australia. W. Raymond is the recipient of a John Donald Stewart Scholarship from the Arthritis Foundation of Western Australia.
- Accepted for publication May 30, 2019.








{kind=link}
{kind=link}