

Abstract
Objective. Antimalarials (AM) are recommended for all systemic lupus erythematosus (SLE) patients without specific contraindications. Their main adverse effect is retinal damage; however, heart disease has been described in isolated cases. The aim of our study is to describe 8 patients with AM-induced cardiomyopathy (AMIC) in a defined SLE cohort.
Methods. Patients attending the Toronto Lupus Clinic and diagnosed with definite (based on endomyocardial biopsy; EMB) and possible AMIC were included [based on cardiac magnetic resonance imaging (cMRI) and other investigations].
Results. Eight female patients (median age 62.5 yrs, disease duration 35 yrs, AM use duration 22 yrs) were diagnosed with AMIC in the past 2 years. Diagnosis was based on EMB in 3 (extensive cardiomyocyte vacuolation, intracytoplasmic myelinoid, and curvilinear bodies). In 4 patients, cMRI was highly suggestive of AMIC (ventricular hypertrophy and/or atrial enlargement and late gadolinium enhancement in a nonvascular pattern). Another patient was diagnosed with complete atrioventricular block, left ventricular and septal hypertrophy, along with concomitant ocular toxicity. All patients had abnormal cardiac troponin I (cTnI) and brain natriuretic peptide (BNP), whereas 7/8 also had chronically elevated creatine phosphokinase. During followup, 1 patient died from refractory heart failure. In the remaining patients, hypertrophy regression and a steady decrease of heart biomarkers were observed after AM cessation.
Conclusion. Once considered extremely rare, AMIC seems to be underrecognized, probably because of the false attribution of heart failure or hypertrophy to other causes. Certain biomarkers (cTnI, BNP) and imaging findings may lead to early diagnosis and enhance survival.
Antimalarials (AM), mainly chloroquine and hydroxychloroquine (HCQ), represent the cornerstone of the longterm management of patients with systemic lupus erythematosus (SLE). Apart from their multiple beneficial effects, their safety profile is quite acceptable. Of the adverse effects, retinopathy is the best known1. About 70% of patients with SLE are administered longterm AM in large cohorts2. Less prevalent adverse effects include neuromyopathy and cardiomyopathy3,4. The latter has been reported only in isolated case reports (47 thus far), with factors such as duration of use and cumulative dose appearing to be decisive for disease development5. However, given the large number of patients treated with AM, it seems possible that this complication is significantly underrecognized. Hypertrophic cardiomyopathy and heart failure, the most common clinical features of AM-induced cardiomyopathy (AMIC), may be falsely attributed to other causes, such as arterial hypertension or ischemic cardiomyopathy. Consequently, nonspecific therapeutic approaches with diuretics and/or antihypertensives will exert minimum or even deleterious effects on such patients.
In the University of Toronto Lupus Clinic, we diagnosed 8 patients with definite and possible AMIC during the past 2 years. The aim of our present report is to describe these patients, with particular emphasis on the diagnostic approach and followup.
MATERIALS AND METHODS
All the patients described herein regularly attend the University of Toronto Lupus Clinic and fulfilled the American College of Rheumatology 1997 revised criteria for SLE classification, or had 3 criteria plus a histopathological proof of the disease6. Cardiological investigations were initiated based on signs and symptoms of heart failure or as a result of abnormal levels of heart biomarkers [cardiac troponin I (cTnI) and/or brain natriuretic peptide (BNP)]. All patients provided written informed consent for studies being conducted in the University of Toronto Lupus Clinic, and the studies are approved by the University Health Network Research Ethics Board (11-0398-AE).
RESULTS
Electrocardiographic, echocardiographic, and cardiac magnetic resonance imaging (cMRI) findings, and disease evolution for all patients, are shown in Table 1.
Demographics, AM duration, ECG, heart imaging, biomarkers, and outcome of AMIC patients.
Patient 1
The original description of the case has previously been published7. Briefly, a 74-year-old female (initial SLE diagnosis at 30) presented with congestive heart failure. AMIC diagnosis was confirmed by endomyocardial biopsy (EMB). She was taking HCQ for 13 years at a dose of 342 mg/day (6.5 mg/kg).
Heart-specific biomarkers (cTnI and BNP) were monitored on a quarterly basis and were steadily decreasing after an initial increase in the first 3 months after HCQ cessation. However, none of them were normalized 2 years after diagnosis (cTnI from 270 to 58 ng/l, normal range < 26 ng/l; BNP from 1080 to 281 pg/ml, normal range < 100 pg/ml). On the contrary, creatine phosphokinase (CPK), which was abnormal for 9 years before AMIC diagnosis, normalized 18 months after drug cessation. The patient is currently asymptomatic; no SLE activity has been observed to date.
Patient 2
A 68-year-old female patient (initial SLE diagnosis at 46) presented with progressive exertional dyspnea and mild pedal edema. She had clinically and serologically inactive disease for many years while taking chloroquine 250 mg/day since diagnosis (2.9 mg/kg). Comorbidities included arterial hypertension, which was tightly controlled (confirmed with 24-h blood pressure monitoring). She had chronically (for 19 yrs) elevated CPK, ranging between 2 and 4 times the upper limit of normal (ULN), with no clinical or electromyographic evidence of myositis. cTnI was 217 ng/l and BNP 382 pg/ml. EMB showed mild interstitial fibrosis, while most myocardial fibers were hypertrophied with marked vacuolation (intracellular lipid accumulation, sarcotubular dilatation). On electron microscopy, most cardiac fibers showed abundant lamellar phospholipid membranes (myelinoid inclusions; Figure 1A) and curvilinear bodies (Figure 1B). Pathologic findings were characteristic of AMIC.
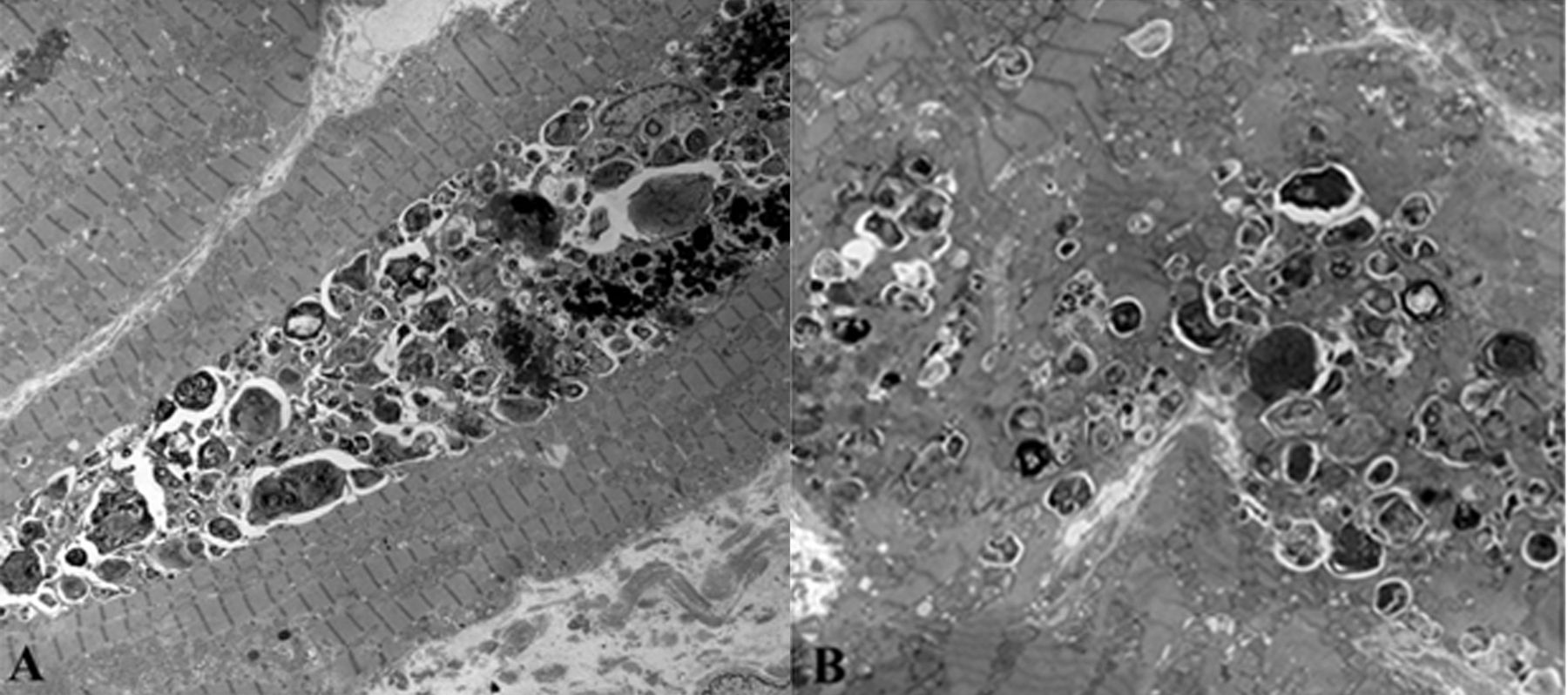
A. Electron microscopy (magnification × 2500). Electron dense, myelin figure–like (myelinoid) inclusions within the myocardial fibers. B. Magnification × 5000. Multiple small electron-dense structures (curvilinear bodies).
Chloroquine had been discontinued 3 months before the biopsy because of high clinical suspicion. However, she developed atrial fibrillation with syncopic episodes interspersed with severe bradycardia, and she received a permanent pacemaker 18 months after chloroquine cessation. She is currently asymptomatic with no SLE activity.
Patient 3
A 65-year-old female patient (initial SLE diagnosis at 12 yrs) with clinically and serologically inactive disease was found to have elevated cTnI and BNP levels at 49 ng/l and 235 pg/ml, respectively. She was completely asymptomatic; prolonged treatment with HCQ (for 45 yrs, 6 mg/kg) prompted testing for the heart biomarkers. Comorbidities included arterial hypertension that was well controlled based on 24-h blood pressure monitoring. Coronary angiography was normal. EMB showed mild interstitial fibrosis and hypertrophic myocardial fibers with marked vacuolation. On electron microscopy, most cardiac fibers showed abundant lamellar phospholipid membranes (myelinoid inclusions). Pathologic findings were characteristic of AMIC.
The patient is currently asymptomatic and closely monitored.
Patient 4
A 59-year-old female (initial SLE diagnosis at 21 yrs with diffuse proliferative nephritis) developed endstage renal disease and received a kidney transplant at the age of 42 years. Comorbidities included arterial hypertension, dyslipidemia, and mitral valve prolapse. Two years ago, the patient developed Takotsubo cardiomyopathy (apical ballooning of the left ventricle), from which she recovered fully after a few months. SLE had been consistently inactive, both clinically and serologically, while taking HCQ 200 mg/day for 19 years (4.1 mg/kg), prednisone 5 mg/day, mycophenolic acid 720 mg/day, cyclosporine 250 mg/day, atorvastatin 10 mg/day, amlodipine 5 mg/day, and metoprolol 25 mg/day.
On regular reassessment of her cardiac condition, she was found to have abnormal heart biomarkers (cTnI 52 ng/l and BNP 182 pg/ml; both were previously normal). Findings were highly suggestive of AMIC and HCQ was discontinued. During followup, heart biomarkers were decreased (cTnI from 52 to 27 ng/l and BNP from 182 to 97 pg/ml) after 6 months. Repeated cMRI at that time showed slight regression of the left ventricular hypertrophy (LV mass index 85 from 90 g/m2).
The patient is currently asymptomatic 15 months after HCQ discontinuation.
Patient 5
A 74-year-old female (initial SLE diagnosis at 47) presented with symptoms of heart failure including exertional dyspnea and bilateral pedal edema. Comorbidities included chronic kidney disease (initially focal proliferative lupus nephritis, baseline eGFR ranging between 30 and 40 ml/min) and arterial hypertension. SLE was clinically and serologically inactive for many years while taking HCQ 400 mg/day (for 24 yrs, 4 mg/kg), nifedipine 30 mg/day, and irbersartan 150 mg/day. She had chronically elevated CPK (for 15 yrs, range 2–4× the ULN) along with skin hyperpigmentation, which was attributed to HCQ.
Her cardiac biomarkers were abnormal (cTnI 139 ng/l and BNP 454 pg/ml). Coronary angiography was normal. Diagnosis of AMIC was made and HCQ was discontinued; however, the patient developed refractory heart failure 3 months after that. In addition, she developed atrial fibrillation, which was managed with catheter ablation and anticoagulation. Subsequently, she developed pulmonary hemorrhage and was admitted to the intensive care unit, where she succumbed to septic shock.
Patient 6
A 54-year-old female (initial SLE diagnosis at 16) had chronically inactive disease under HCQ 400 mg/day for 22 years (dose 6.6 mg/kg). Comorbidities included mild coronary artery disease (solitary occlusion of 20% in the right coronary artery) that was managed conservatively. Her CPK was chronically elevated, ranging between 2 and 4 times the ULN. HCQ was discontinued a few months ago because of retinal toxicity. The patient was completely asymptomatic; prolonged AM treatment and elevated CPK, along with AM-related ocular toxicity prompted further testing. Her cTnI was 174 ng/l and BNP 85 pg/ml. Followup after 3 months (HCQ had not been discontinued yet) showed the same levels of cTnI (180 ng/l) and abnormal BNP (155 pg/ml). Nine months after drug cessation, cTnI was 90 ng/l and BNP 121 pg/ml.
The patient is currently asymptomatic 12 months after drug cessation.
Patient 7
A 49-year-old female (initial SLE diagnosis at 17 yrs) had clinically and serologically inactive disease for many years under chloroquine 250 mg/day (for 23 yrs, dose 2.6 mg/kg). Comorbidities included arterial hypertension (well-controlled with atenolol 50 mg/day and lisinopril 20 mg/day). Her CPK was elevated and ranging between 2 and 4 times the ULN for the last 14 years. The patient was completely asymptomatic; prolonged AM treatment and elevated CPK prompted further testing. Her cTnI was 75 ng/l and BNP 122 pg/ml.
AMIC was suspected and chloroquine was discontinued. During followup, heart biomarkers were monitored on a quarterly basis and decreased steadily (cTnI from 75 to 28 ng/l, and BNP from 122 to < 10 pg/ml after 18 mos). CPK levels were normalized 15 months after drug cessation.
The patient flared (polyarthritis) 18 months after chloroquine cessation and was treated with methotrexate.
Patient 8
A 60-year-old female (initial SLE diagnosis at 51 yrs) had chronically quiescent disease under HCQ 400 mg/day for 9 years (dose 7.9 mg/kg). Her CPK was chronically elevated. She developed retinal toxicity and HCQ was discontinued. A week later, she developed complete atrioventricular block and received a permanent pacemaker. Her cTnI was elevated at 81 ng/l and BNP at 184 pg/ml. Coronary angiography was normal. During followup, there was a regression of left ventricular hypertrophy after 9 months; heart biomarkers were decreased (cTnI from 81 to 42 ng/l, and BNP from 184 to 97 pg/ml).
The patient is currently asymptomatic under no treatment for her SLE.
DISCUSSION
AMIC is considered extremely rare in the clinical setting, having been reported in 47 sporadic cases thus far5. In those patients, the median AM treatment duration was 12 years5, while our patients were receiving AM for a median 22 years. The cumulative dose was 1005 g for chloroquine (n = 22) and 1542 g for HCQ (n = 22); in our series, these numbers were significantly larger (2055 g and 2419 g, respectively). The optimal duration of AM treatment in patients with SLE has not been determined yet. Consequently, the majority of them remain on AM indefinitely, therefore increasing the risk for cardiomyopathy.
Clinical presentation was most commonly that of congestive heart failure (77%) with dyspnea of varying severity and peripheral edema, whereas syncope and/or presyncopal episodes manifested in 17% of the patients. In our series, 3 patients were diagnosed during investigation of new-onset heart failure (2 with preserved left ventricular ejection fraction) and 1 with syncope. In the remaining 4 patients, abnormal heart biomarkers, specifically cTnI and BNP, triggered further testing. In a systematic literature review5, cTnI was reported abnormal in 8 patients with AMIC7–14 and normal in 115; interestingly, 1 patient had intermittently abnormal troponin for 2 years before diagnosis13. BNP was found abnormal in all 10 cases where it was reported7,10,12–18. All our patients had both biomarkers elevated at the time of diagnosis, even with no symptoms. Further, their levels were gradually decreased over time after drug cessation, thus providing a tool for monitoring such patients. However, the rate of decrease was rather slow, probably reflecting the prolonged half-life and the large distribution volume of AM19.
From a heart structural perspective, left ventricular hypertrophy was detected in all our patients, while right ventricular hypertrophy in half of them. In the literature, ventricular hypertrophy was reported in 74.3% and 51.4%, respectively5. Impaired systolic function (defined as a left ventricular ejection fraction < 50%) was found in only 1 of our patients (52.8% in the literature), probably because most of our patients were completely asymptomatic at the time of investigation. It seems possible that AMIC is a chronic process and systolic dysfunction will become apparent only in late stages. Diastolic dysfunction, however, may be an early finding. Restrictive filling pattern of the left ventricle was documented in 51.4% of the published cases, while it was apparent in 7 of our patients.
Interestingly, hypertrophy of the interventricular septum was observed in all our patients; it was mild to moderate with a septal thickness between 12 and 16 mm (normal < 11 mm). The same findings have been reported in 43% of the published AMIC cases5. Septal hypertrophy was confirmed with cMRI in 7 of our patients; in the eighth, it was not performed owing to her pacemaker. In addition, late gadolinium enhancement (LGE) was reported in 4 patients with a patchy, nonvascular pattern distribution in the interventricular septum and the lateral wall of the left ventricle. The significance of these findings is not clear. Experimental data from animals have shown heart damage primarily in the septum following chloroquine administration20. Further, the existence of melanocytes in the heart has been shown for both animals and humans; their distribution was in the interventricular septum and around the atrioventricular valves21. AM show a high affinity for melanocytes and this may lead to a selective deposition in these cells.
cMRI is considered the imaging modality of choice in nonischemic cardiomyopathies. However, conventional techniques failed to reveal any specific findings. In most cases, it confirmed the echocardiographic findings, whereas it also showed LGE in a nonvascular pattern in about half the cases. This is related to myocardial fibrosis and scar tissue, although it is unknown whether it represents a reparative process following AM-induced myocardial necrosis22. In inflammatory myocarditis, a peculiar LGE pattern along with edema is usually detected23. Ischemic and nonischemic cardiomyopathies can be reliably differentiated; however, the extent or distribution of LGE cannot characterize the precise type of fibrosis. Novel techniques, such as T1 mapping and quantification of the extracellular volume, may overcome this weakness23. In Anderson-Fabry cardiomyopathy, which resembles AMIC in the pathophysiological and histopathological level, this technique detected subclinical heart damage, even before the development of cardiac hypertrophy24.
This predilection of AM for deposition in the interventricular septum may be the reason for the increased prevalence of conduction abnormalities in such patients. Half of our patients had right bundle branch block and 2 of them had bifascicular block; in 1 case, complete atrioventricular block initiated investigations. In the literature, about half of the patients with AMIC manifested complete atrioventricular block and received a permanent pacemaker, occasionally up to 4 years before diagnosis5. About 25% of the patients were reported to have right bundle branch block and 14% left bundle branch block5. In the general population, the prevalence of bifascicular block (right bundle branch and left anterior fascicular block) is reported to be 1%, while it increases to 25% in patients with recurrent syncope25. This condition carries a 50% probability to evolve into a complete atrioventricular block 5 years from diagnosis26. In cases with AMIC and complete block, the progress of the conduction disorder was characterized by the development of right bundle branch block, and subsequently, left anterior fascicular block months or years before the complete block and histopathological diagnosis4,5.
In this series, we also observed that most patients had elevated CPK for years before the development of cardiomyopathy. That ranged between 2 to 4 times the ULN in all patients with no clinical or electromyographic evidence of myositis or myopathy. Apart from 1 patient who was concomitantly treated with statins, abnormal CPK had been attributed to prolonged AM treatment. We have previously shown that AM confer a 3-fold increased risk for chronic CPK elevation in patients with SLE, even after excluding patients taking statins27. The precise pathophysiologic link between muscle damage and AMIC is not known. It is possible that drug deposition within the myocytes is not limited to skeletal muscles; cardiomyocytes may be affected as well. In this context, skeletal muscle biopsy was performed in 4 cases and showed findings similar to the EMB (vacuolation of myocytes and curvilinear bodies)5. About one-fourth of the published cases had clinical myopathy at the time of diagnosis, while CPK levels were elevated in almost 80% of the cases, probably reflecting AM-related subclinical muscle toxicity28.
The mortality rate of AMIC is estimated at 45%; death occurred within 3 months after diagnosis on average5. Another 2 patients received heart transplants shortly (3 and 4 mos) after diagnosis29,30. In our series, only 1 patient died (12.5%). This better survival may be due to the early diagnosis in these patients.
The reversibility of AMIC remains controversial. In the published cases of AMIC, congestive heart failure was clinically and functionally improved (as assessed by the left ventricular ejection fraction) in half the patients5. Evidence of regression of ventricular hypertrophy or atrial dilatation was provided by echocardiography in almost half the patients as well5. On the contrary, about 15% of the patients did not show any evidence of regression, even after 2 years31,32,33. Moreover, in both patients managed with heart transplantation, AM had been discontinued 3 and 4 months, respectively; however, no hypertrophy regression or functional improvement was noted29,30. In our patients, regression of ventricular hypertrophy was observed after 6–9 months in cMRI in 4 out of 5 patients in whom it was performed. However, we did not observe any regression in septal hypertrophy. In addition, in patients with conduction abnormalities there were no changes; on the contrary, in 2 patients, conduction damage continued to progress. Given that drug deposition in the tissues may act as a reservoir and lead to further damage, it seems reasonable that hypertrophy regression will be a slow process. In this context, it has been shown that chloroquine can be detected in the urine for years after drug withdrawal4, while retinal damage frequently progresses after stopping AM34.
AMIC is an underrecognized complication of prolonged AM treatment. It presents as a hypertrophic, restrictive cardiomyopathy with or without conduction abnormalities. Patients taking prolonged AM treatment, and particularly those with chronically elevated CPK, are at risk and should be monitored closely. Heart-specific biomarkers, such as cTnI and BNP, may serve as a screening tool for detection of myocardial injury and should be monitored in regular time intervals. Electro- and echocardiography may provide further evidence of subclinical conduction abnormalities and/or structural impairment (septal hypertrophy, diastolic dysfunction, hypertrophy). More thorough investigations, such as cMRI, seem justifiable in patients with positive findings. Heart biopsy remains the gold standard for diagnosis by the detection of cardiomyocyte vacuolation on light microscopy and lamellar and curvilinear bodies ultrastructurally (representing autophagolysosomes with improperly digested cellular organelles). However, drug cessation should be prompt and probably upon suspicion of AMIC, because complete investigation may be delayed significantly.
Footnotes
K. Tselios is financially supported by the Geoff Carr Fellowship from Lupus Ontario. The University of Toronto Lupus Research Program is supported by the University Health Network, Lou and Marissa Rocca, and the Lupus Foundation of Ontario.
- Accepted for publication July 24, 2018.








{kind=link}