

To the Editor:
Primary immunodeficiency diseases (PID) are a heterogeneous group of diseases with variable genetic etiologies. Although immunodeficiency is a hallmark of susceptibility to infection, autoimmunity is clearly a prevalent feature1. Some PID, including common variable immunodeficiency (CVID), are often adult-onset conditions2. Heterozygous gain-of-function (GOF) mutations in the signal transducer and activator of transcription 1 (STAT1) gene have been identified as underlying causes of chronic mucocutaneous candidiasis, a childhood-onset PID3; however, its comprehensive clinical features and outcomes remain undefined. We present a patient with a STAT1 GOF mutation associated with multiple autoimmune diseases, including Takayasu arteritis (TA), a previously unreported phenotype of STAT1 GOF mutations.
We obtained ethics approval for our study from the ethical committee of the Faculty of Medicine, Oita University, Japan (ethics approval number: 1387). Written informed consent was obtained from the patient’s family to publish the material.
The patient was a 25-year-old Japanese woman diagnosed at 8 years of age with autoimmune hypothyroidism and treated with hormonal substitution therapy. She lacked a familial history of associated PID and remained symptom-free throughout childhood and adolescence under hormonal therapy. At 21 years, TA was accidentally diagnosed because of a pulseless right radial artery, with no other symptoms. TA-associated HLA-B674 was positive. Damaged large vessels from neck to lower limbs (Figure 1) and severe aortic regurgitation developed; patient underwent aortic valve replacement at 22 years. She was kept on a corticosteroid and tacrolimus regimen to maintain a low inflammation level, but severe inflammatory bowel disease (IBD) developed at 23 years. The patient favorably responded to combined methotrexate and infliximab (IFX) instead of tacrolimus. However, her IgG levels gradually declined to 400–600 mg/dl; she required multiple hospitalizations for various infections such as cellulitis, herpes zoster, genital herpes, oral candidiasis, genital abscess, and Pneumocystis carinii pneumonia at 25 years. She was simultaneously receiving prednisolone therapy (PSL) at 9 mg/day; we suspected secondary immunodeficiency and suspended all immunosuppressive drugs except PSL. After 4 months, IBD reappeared and we observed partial clinical amelioration after increasing PSL dose to 30 mg/day. Further hypogammaglobulinemia (IgG < 300 mg/dl) and B cell, but not T cell, lymphopenia (CD19+ cells: 0.1%; normal range: 6–23%) emerged. She was hospitalized for serious melena associated with multiple ulcers in the upper gastrointestinal (GI) tract. We reduced PSL dosage and reintroduced IFX; this partially restored gamma globulin levels and ameliorated GI lesions. Unexpected rapid progressive thrombocytopenia occurred, requiring daily platelet transfusions. Although high-dose PSL effectively treated the thrombocytopenia, indicating an autoimmune component of the underlying condition, reduced PSL dosage was necessary for the infections. She was diagnosed with intractable PID based on medical history, clinical presentation, and laboratory test results including hypogammaglobulinemia with B cell deficiency and marked reduction in regulatory T cells (Treg) and interleukin (IL)-17A-producing CD4+ T cells percentages (Figure 2A and 2B). The potential treatment with hematopoietic stem cell transplant was difficult because of the significant risk to our patient. We prescribed cyclosporine, abatacept, and tacrolimus but obtained minimal effects. Our patient died from a diffuse alveolar hemorrhage of uncertain origin; an autopsy was not performed. Using DNA sequencing, we identified a previously recognized STAT1 GOF mutation, c.970T>C, p.C324R, in the DNA binding domain (Figure 2C)5,6. DNA sequencing revealed no evidence of CVID- or other candidate PID-associated genetic alterations in any of 44 candidate genes tested (including STAT3, CTLA4, AIRE, and FOXP3). We diagnosed a PID due to a STAT1 GOF mutation accompanied by B cell and Treg deficiencies and hypogammaglobulinemia, which are rare but possible immunological features of the disease7.

Images of CT angiography at diagnosis of Takayasu arteritis at the age of 21 years. CT: computed tomography.
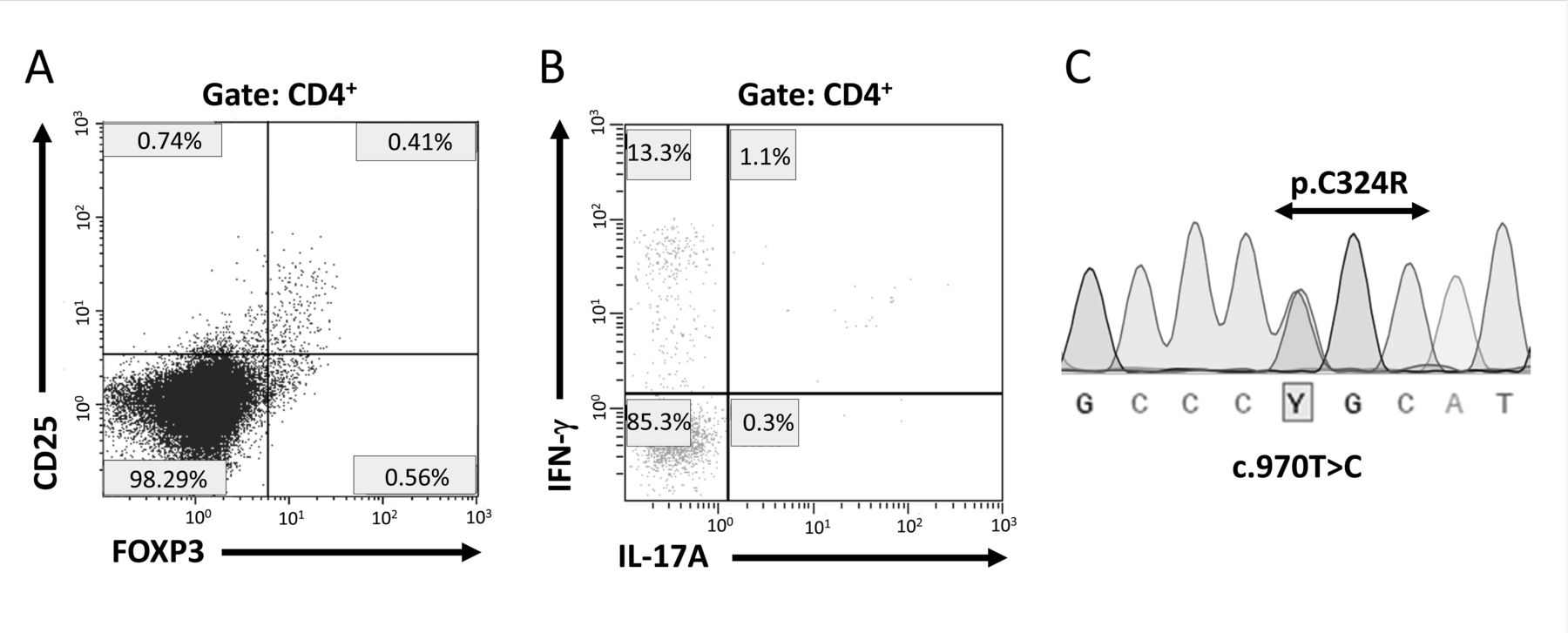
A. Flow cytometric analysis of peripheral blood from the patient demonstrating low percentage of regulatory T cells (normal values, FoxP3+ of CD4+: 4–8%). The populations of CD4/8, naive, and memory T cells were normal. B. Flow cytometric analysis of IL-17A and IFN-γ secretion by peripheral CD4+ cells stimulated with phorbol myristate acetate/ionomycin for 4 h prior to intracellular staining (normal values, IL-17+ of CD4+: 6–22%). C. Sanger DNA sequencing chromatogram of the mutated STAT1 gene. Protein-coding exons and their intronic boundaries of 45 genes, including STAT1, were sequenced as PCR products on an ABI 3130/3730 capillary sequencer (Thermo Fisher Scientific) and/or a MiSeq next-generation sequencer (Illumina). IL: interleukin; IFN: interferon.
Hypothyroidism, IBD, and thrombocytopenia are common autoimmune features of STAT1 GOF mutations3. A few large vessel aneurysm cases with STAT1 GOF mutations have been reported3,8; however, to our knowledge, this is the first case reporting TA as a complication. Although TA onset in this patient could be coincidental and independent of STAT1 GOF mutation, the STAT1 signaling pathway activation can amplify the cellular responses to type I and II interferons in patients harboring a STAT1 GOF mutation9. Interferon-γ is involved in TA pathophysiology10, indicating a possible association between TA and the STAT1 GOF mutation in our patient. The presence of HLA-B67 may have also modified the phenotype. Most patients with STAT1 GOF mutations exhibit childhood-onset disease3; hypothyroidism occurred at 8 years in our patient. However, most of her STAT1 GOF mutation-related clinical symptoms initiated at adulthood, and immunosuppressive therapy was prescribed before increased susceptibility to infection developed. Thus, PID diagnosis, including STAT1 GOF mutations, was challenging and we could have missed the diagnosis if marked hypogammaglobulinemia was not detected during high-dose PSL therapy.
Rheumatologists should consider the possibility of PID patients exhibiting autoimmune characteristics. Atypical phenotypes for autoimmune diseases, including marked hypogammaglobulinemia, a rare combination of multiple autoimmune diseases, and a history of childhood autoimmunity may be indicators for PID diagnosis, even without susceptibility to infection. A definitive PID diagnosis, defined by single-gene inborn errors of immunity, is important for individualized optimal treatment; therefore, patients presenting with primarily autoimmune features should also be considered for PID diagnosis.
Acknowledgment
We thank the patient and her family for their collaboration. We also thank the entire paramedical and medical staff of Oita University Hospital, as well as Dr. Chiharu Imada and Dr. Yasuhiro Kiyonaga, for their dedicated care. Finally, we are deeply grateful to Dr. Kohsuke Imai, Dr. Hirokazu Kanegane, Dr. Satoshi Okada, Dr. Hidenori Ohnishi, Dr. Osamu Ohara, Dr. Masataka Ishimura, and Dr. Hidetoshi Takada, who are members of the Primary Immunodeficiency Database in Japan, for their continuous excellent support of the study, including genetic and immunological analyses.








{kind=link}
{kind=link}