

Abstract
Objective. Shared epitope (SE) alleles are the most significant genetic susceptibility locus in rheumatoid arthritis (RA); however, their target populations in CD4+ T cells are not well elucidated. We analyzed the association between SE alleles and the T cell receptor (TCR) repertoire diversity of naive and memory CD4+ T cells using next-generation sequencing (NGS).
Methods. The TCR beta chains in naive and memory CD4+ T cells from the peripheral blood of 22 patients with RA and 18 age- and sex-matched healthy donors (HD) were analyzed by NGS. The Renyi entropy was used to evaluate TCR repertoire diversity and its correlations with SE alleles and other variables were examined. Serum cytokine levels were measured by multiplex ELISA.
Results. The TCR repertoire diversity in memory CD4+ T cells was reduced in SE allele-positive patients with RA compared with HD, and showed a significant negative correlation with the SE allele dosage in RA. The TCR repertoire diversity of naive and memory T cells was also negatively correlated with disease activity, and the SE allele dosage and disease activity were independently associated with reduced TCR repertoire diversity. TCR repertoire diversity showed a significant positive correlation with the serum interleukin 2 levels.
Conclusion. SE alleles and disease activity were negatively correlated with the TCR repertoire diversity of CD4+ T cells in RA. Considering the pivotal role of CD4+ T cells in RA, restoring the altered TCR repertoire diversity will provide a potential RA therapeutic target.
Rheumatoid arthritis (RA) is characterized by chronic synovitis and joint deformity, and the pathogenesis of RA is based on genetic risks and environmental factors1. HLA-DRB1 risk alleles are the most significant genetic susceptibility locus in RA, and these alleles share consensus sequences in the third hypervariable region of the HLA-DRB1 chain, p70-74 (shared epitope; SE)1,2. Structural analysis provided the molecular basis of the SE hypothesis, which states that SE form a determinant position in the antigen-binding P4 pocket. Indeed, some autoantigen-derived peptides are favorably presented on HLA-DRB1*0401 and *0405 molecules3. Therefore, HLA-DRB1 risk alleles represent important roles in antigen presentation and T cell immunity in RA. Indeed, oligoclonal expansion of peripheral and synovial-infiltrating CD4+ T cells suggests the important roles of antigen-specific clonal T cell responses in RA4,5. However, the detailed roles of SE in CD4+ T cells have not been elucidated in RA.
Previous studies reported altered T cell receptor (TCR) repertoires in patients with RA, and showed that TCR repertoire diversity is reduced in RA6,7,8. However, the clinical and pathological significance of this reduced TCR repertoire diversity remains unclear. Herein, we analyzed the association between SE alleles and the TCR repertoire diversity of naive and memory CD4+ T cells from patients with RA and healthy donors (HD) using next-generation sequencing (NGS). Moreover, we collected the exact same number of cells using FACS and analyzed the same number of sequencing reads by random resampling to avoid bias. This comprehensive and unbiased analysis shed light on the close association between clinical variables, including SE alleles and disease activity, and TCR repertoire diversity in RA.
MATERIALS AND METHODS
Patients and HLA typing
Blood samples were obtained from patients with RA (n = 22) and age- and sex-matched HD (n = 18). Patients with RA fulfilled the classification criteria of the 2010 American College of Rheumatology/European League Against Rheumatism9. The demographic and clinical backgrounds of the subjects are summarized in Table 1. There was no statistically significant difference in age according to SE status or between the HD and patients with RA. Clinical information, including the 28-joint count Disease Activity Score using C-reactive protein (DAS28-CRP), Health Assessment Questionnaire–Disability Index, and anticyclic citrullinated peptide (anti-CCP) 2 antibody, was collected along with the blood samples. We defined anti-CCP2 antibody positivity as a titer > 5 U/ml. HLA-DRB1 genotyping of the patients with RA was performed using PCR sequencing–based typing. The HD were genotyped using Infinium OmniExpressExome (Illumina) in accordance with a previous report that assessed the fidelity of imputation10. Written informed consent was obtained from all subjects before sample collection in accordance with the latest version of the Declaration of Helsinki. This study was approved by the Ethical Committee of the University of Tokyo Hospital (10154 and G3582). The methods were performed in accordance with the approved guidelines.
Demographic and clinical backgrounds of the patients with RA, and the HD.
CD4+ T cell sampling
Peripheral blood samples were collected from the HD and patients with RA. Peripheral blood mononuclear cells (PBMC) were isolated by density-gradient centrifugation using Ficoll-Paque PLUS (GE Healthcare). After treatment with an Fc receptor–binding inhibitor (eBioscience), PBMC were stained with the following antibodies: anti-human CD3 (UCHT1, Biolegend), anti-human CD4 (OKT4, Becton, Dickinson and Co.), and anti-human CD45RA (HI100, Biolegend) antibodies. Naive (CD3+CD4+CD45RA+) and memory CD4+ T cells (CD3+CD4+CD45RA-) were sorted using the Moflo XDP cell sorter (Beckman Coulter), and up to 50,000 cells from each population, with purities of > 99.5%, were used for following RNA extraction (Supplementary Figure 1, available with the online version of this article).
Preparation of cDNA library
We used our method of cDNA library preparation reported previously, with minor modifications11,12. Briefly, RNA was extracted from sorted cells using the RNeasy micro kit (QIAGEN) and reverse transcribed from total amounts of the extracted RNA samples using Superscript III (Invitrogen) and the gene-specific primers, hTCR-CB1-R3.2 and TCR-CB2-R3 (primer sequences are listed in Supplementary Table 1, available with the online version of this article). The resulting cDNA was amplified using 5-min rapid amplification cDNA end PCR strategy, which enabled amplification of the upstream cDNA sequences by inverse PCR to obtain the TCR beta CDR3 sequence without using specific primers targeting the V regions (Supplementary Figure 2, available with the online version of this article).
NGS and TCR repertoire analysis
Amplified cDNA was fragmented using the Covaris S2 sonicator. Sequencing adaptors were ligated to the cDNA fragments using the KAPA library preparation kit (KAPA). Paired-end sequencing was performed using the Miseq Reagent V2 kit (Illumina) on the MiSeq system. All procedures were performed following the instructions provided with the kits.
The median read count per sample was 749,702 (range 505,832–918,286). Sequences containing adaptors or low-quality bases (Phred quality score < 20) were removed. Each read was mapped to the TCR genes, and CDR3 sequences were determined using MiXCR13. About 20% of all reads mapped to CDR3 sequences (range 101,821–230,168). Representative cumulative plots are indicated in Supplementary Figure 3 (available with the online version of this article). Because these curves nearly reached a plateau, we considered the TCR sequencing performed in our study to be exhaustive. To avoid the bias induced by the different read counts among samples, we randomly resampled 100,000 reads with coding potential in each sample. The frequencies of each unique clone were used to estimate diversity. We used the Renyi entropy for estimation of TCR repertoire diversity7,8. The Renyi entropy is a comprehensive index used to evaluate TCR repertoire diversity according to the extent of clonal expansion and distribution, and is dependent on the variable α. For example, when α > 1, more weight is placed on highly expanded clones, and when α < 1, more weight is placed on rarely expanded clones7,8. To analyze correlations, we assessed TCR repertoire diversity using the Renyi entropy with α = 1, also referred to as the Shannon index7,8. The concordance rate of the Renyi index was > 99.5% between biological replicates (Supplementary Figure 4, available with the online version of this article).
Serum cytokine multiplex ELISA
Serum cytokine concentrations were measured using customized multiplex ELISA (MILLIPLEX Multiplex Assay, Millipore). Five cytokines [interleukin (IL)-2, IL-6, IL-7, IL-15, and tumor necrosis factor-α] were selected for evaluation.
Statistical analysis
Statistical analysis was performed using computer software R (version 3.2.1). The Tukey-Kramer method was used for comparisons of continuous variables among > 3 groups. Spearman correlation coefficient was used to demonstrate the association between 2 variables. The Renyi entropy was used to evaluate TCR repertoire diversity (vegan, R package). To analyze the independent predictors of TCR repertoire diversity, multiple linear regression analysis was performed. P < 0.05 was considered to indicate statistical significance.
RESULTS
SE allele dosage was significantly and independently correlated with the reduced TCR repertoire diversity of CD4+ T cells in RA
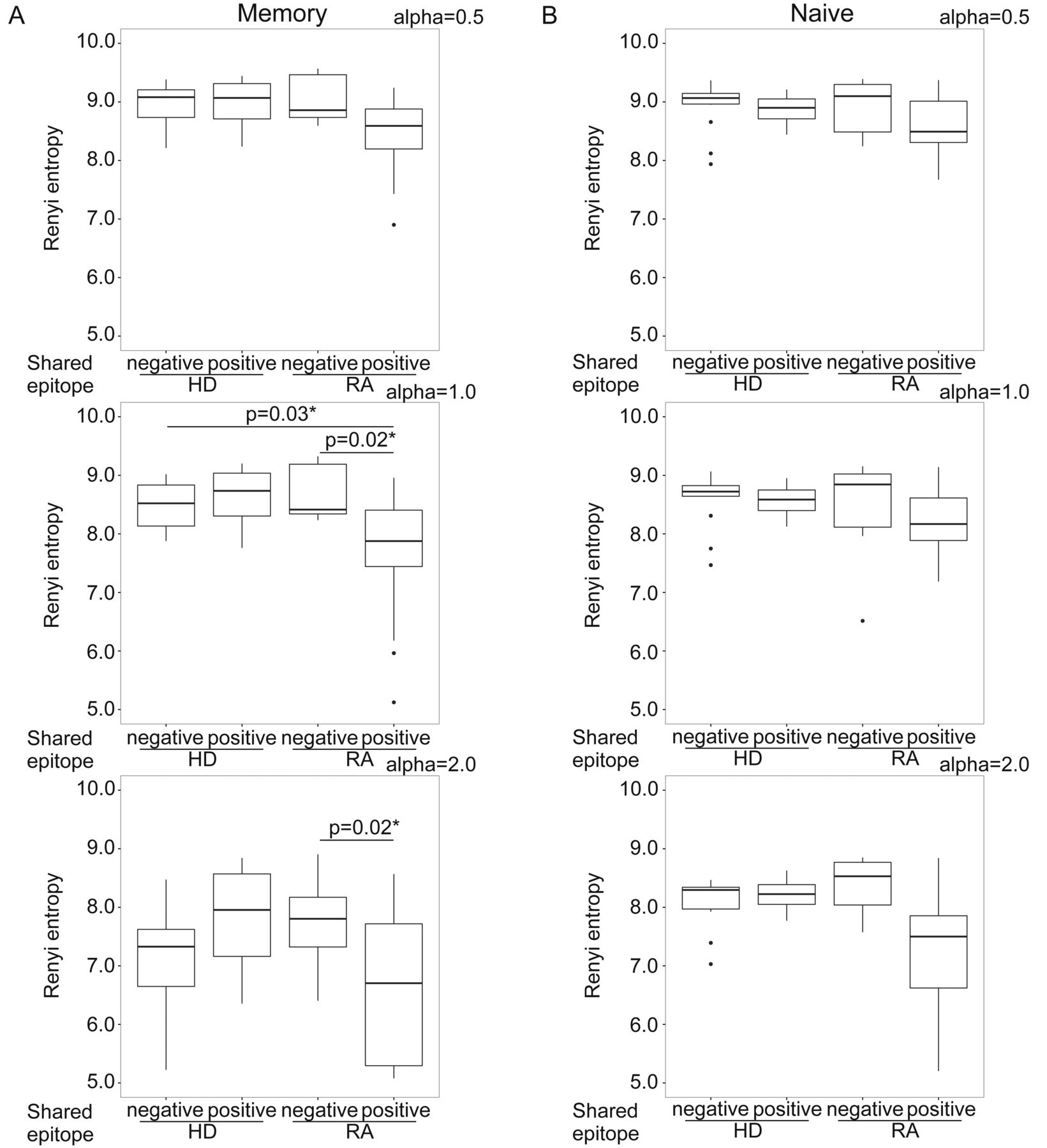
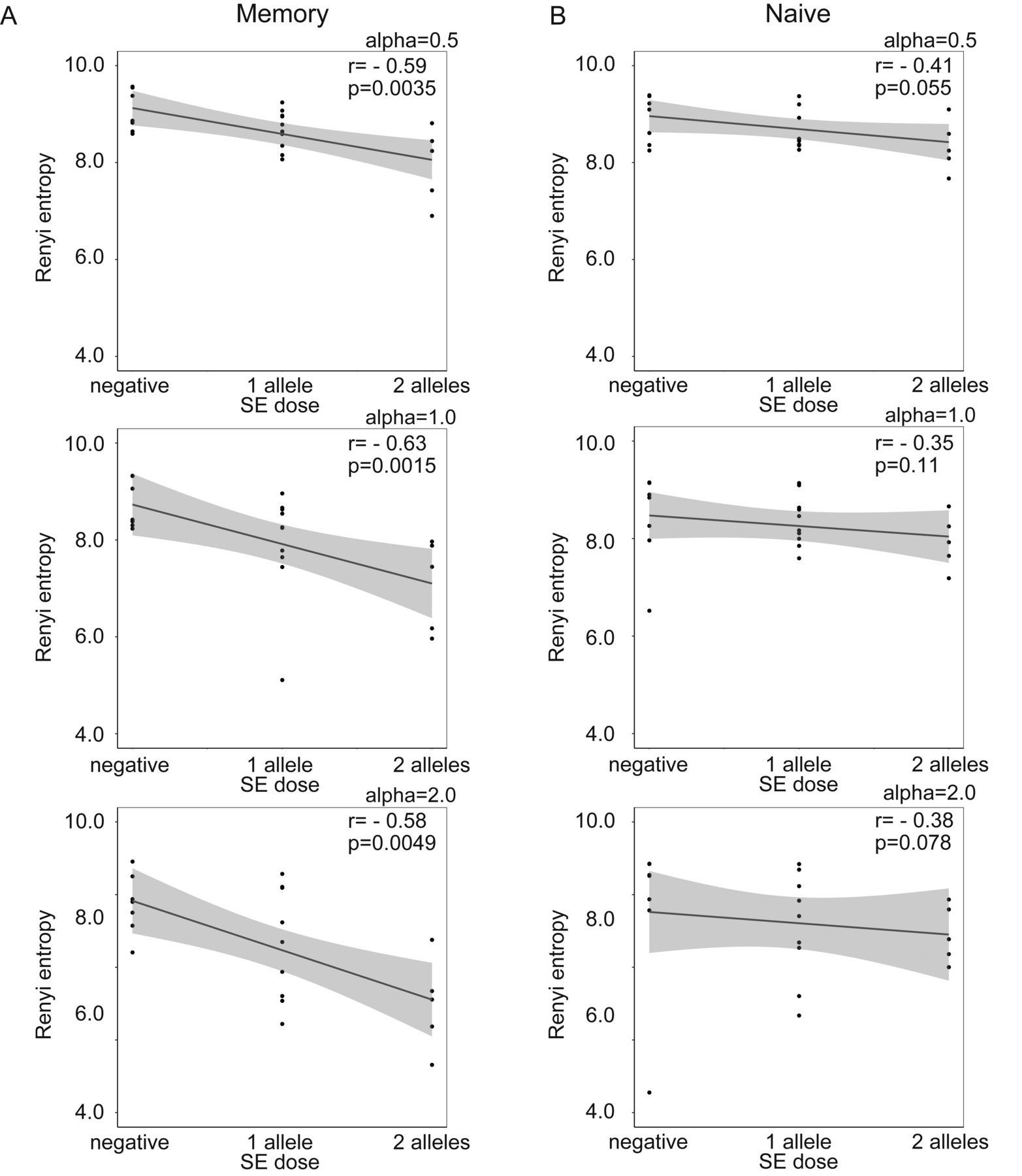
First, the relationship between SE alleles and TCR repertoire diversity was evaluated in naive and memory CD4+ T cells. The TCR repertoire diversity of memory CD4+ T cells, when evaluated using the Renyi index with α > 1, was significantly reduced in SE allele-positive patients with RA compared with SE allele-negative patients with RA and SE allele-negative HD (Figure 1A). The TCR repertoire diversity of memory CD4+ T cells, when evaluated using the Renyi index α < 1, showed a decreasing tendency in SE allele-positive patients with RA (Figure 1A) but did not differ between SE allele-positive and -negative HD (Figure 1B). Moreover, the TCR repertoire diversity of memory CD4+ T cells showed a significant negative correlation with the SE allele dosage in patients with RA (Figure 2A). The TCR repertoire diversity of naive CD4+ T cells showed a slight decreasing tendency in SE allele-positive patients with RA, although this was not significant (Figure 1). A weak correlation was also observed between the SE allele dosage and TCR repertoire diversity of naive CD4+ T cells in patients with RA (Figure 2B). These observations suggested that SE alleles were associated with the reduced TCR repertoire diversity, especially in memory CD4+ T cells of patients with RA, in a dose-dependent manner. Regarding the public clonotypes, the previously identified RA-associated public clonotypes were examined in our samples14. Among these clonotypes, 1 clone (CDR3 sequence YFC ASS LGR NTE AFF GQG) was shared among 4 different patients with RA, even though it had a low abundance (read counts 5, 3, 1, and 2, respectively). However, we rarely detected proliferation or accumulation of the other public clones in SE allele-positive patients.

TCR repertoire diversity of (A) memory and (B) naive CD4+ T cells calculated using the Renyi entropy in SE allele-positive and -negative patients with RA (n = 22) and in age- and sex-matched HD (n = 18), using the representative α index. A detailed explanation of the Renyi entropy is provided in the Materials and Methods section. Statistical analysis was performed using the Tukey-Kramer method. * p < 0.05. TCR: T cell receptor; SE: shared epitope; RA: rheumatoid arthritis; HD: healthy donors.
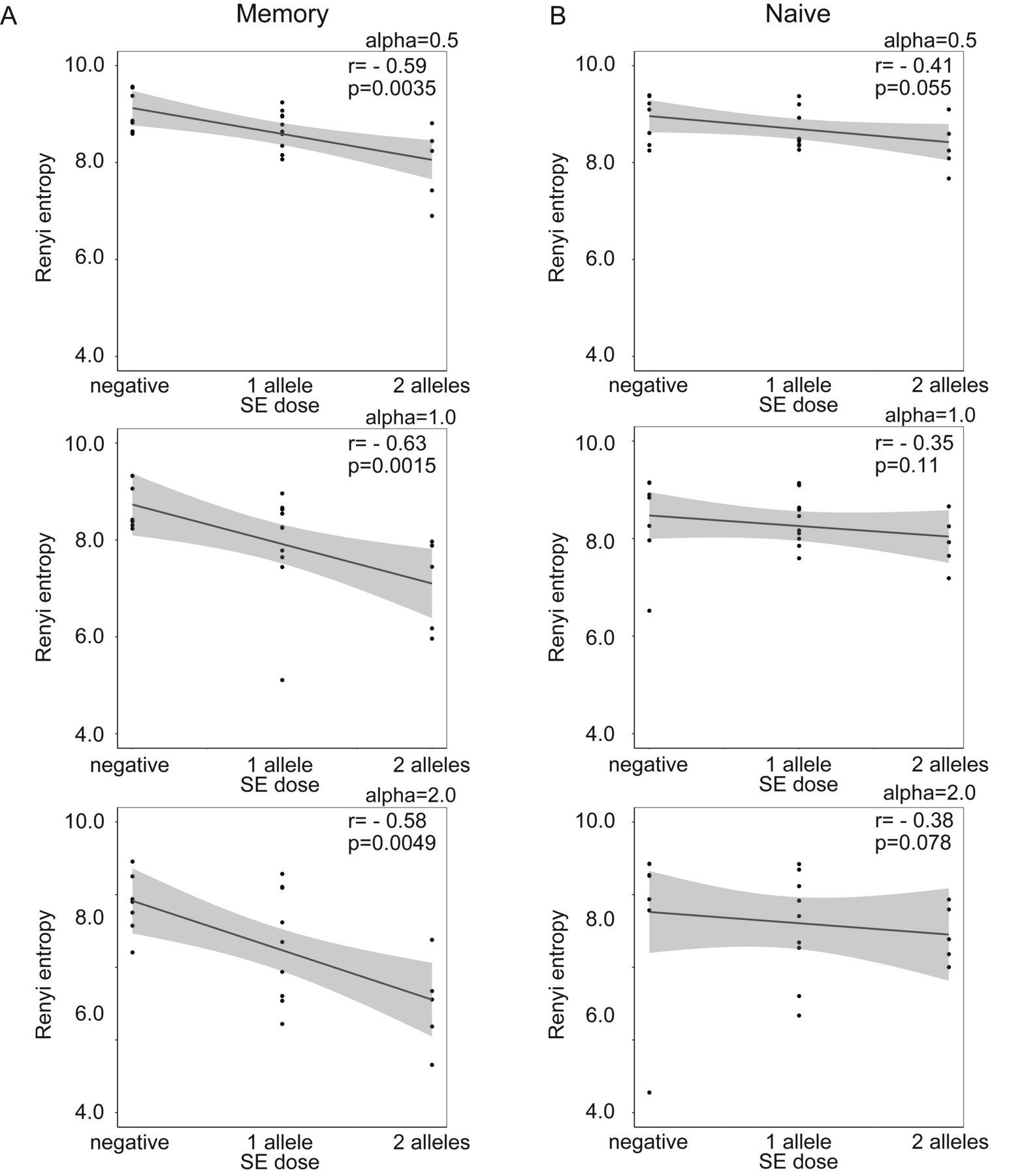
Spearman correlation analysis between SE alleles and TCR repertoire diversity of (A) memory and (B) naive CD4+ T cells in patients with RA, calculated using the Renyi entropy (α = 1). * p < 0.05. SE: shared epitope; TCR: T cell receptor; RA: rheumatoid arthritis.
Next, the correlations between clinical variables and TCR repertoire diversity were analyzed. According to univariate analysis, the SE allele dosage was most significantly associated with the TCR repertoire diversity of memory CD4+ T cells (Figure 3A). The disease activity (as measured by the DAS28-CRP) of RA was also significantly correlated with the TCR repertoire diversity of both memory and naive CD4+ T cells (p < 0.01; (Figure 3A and 3B). This correlation was also observed when the TCR repertoire diversity was evaluated using the Renyi entropy with α > or < 1. In contrast, the anti-CCP2 antibody titers did not show a significant correlation with the TCR repertoire diversity (Figure 3A, B). In addition, the TCR repertoire diversity was not significantly different between anti-CCP2 antibody-positive and -negative patients (Supplementary Figure 5, available with the online version of this article). To infer the independence of the univariate analysis, we assessed the correlations between SE alleles and other clinical variables (Figure 4). The results suggested that the association between the SE allele dosage and TCR repertoire diversity was independent of the disease activity index, DAS28-CRP. Of note, multiple linear regression analysis using DAS28-CRP and SE allele dosage as explanatory variables showed that both were independent predictors of the TCR repertoire diversity of memory CD4+ T cells (p = 0.0297 and 0.0399, respectively).
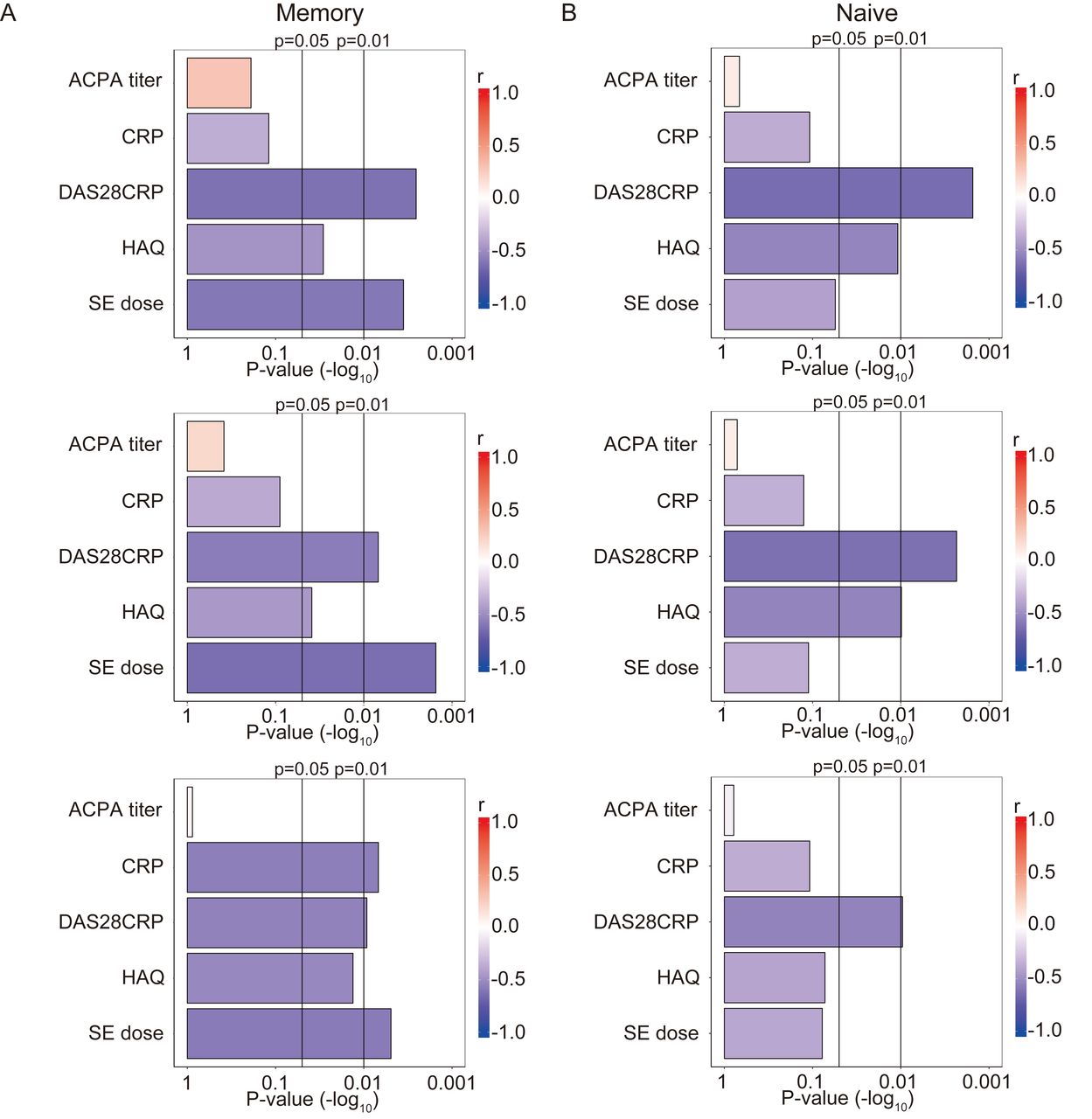
Correlations between the TCR repertoire diversity of (A) memory and (B) naive CD4+ T cells and clinical variables with RA-associated clinical and serological variables in patients with RA (n = 22). P values (−log10) are indicated, obtained by Spearman correlation analysis of TCR repertoire diversity and clinical variables. TCR: T cell receptors; RA: rheumatoid arthritis; ACPA: anticitrullinated protein antibodies; CRP: C-reactive protein; DAS28CRP: 28-joint count Disease Activity Score using CRP; HAQ: Health Assessment Questionnaire; SE: shared epitope.

Correlations between RA-associated clinical variables and the SE allele dosage in patients with RA (n = 22). Rho values indicate the correlations between variables, as determined by Spearman correlation analysis. RA: rheumatoid arthritis; SE: shared epitope; ACPA: anticitrullinated protein antibodies; CRP: C-reactive protein; DAS28CRP: 28-joint count Disease Activity Score using CRP; HAQ: Health Assessment Questionnaire.
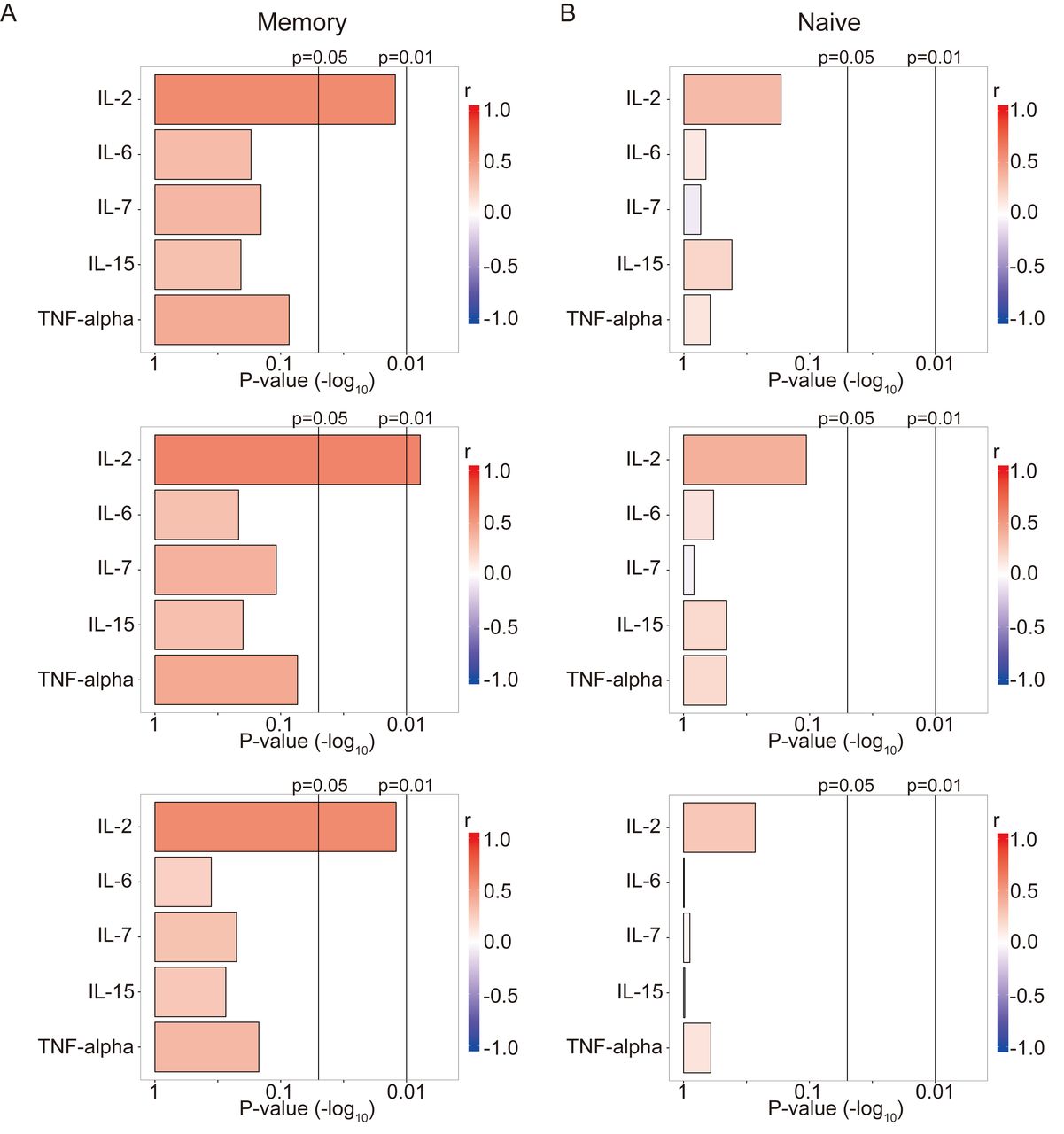
Serum IL-2 concentration was positively correlated with TCR repertoire diversity in RA
Because T cell proliferation, including homeostatic expansion, is reported to play an important role in shaping the TCR repertoire6,15, the correlations between TCR repertoire diversity and serum concentrations of cytokines, including IL-2 and IL-7, were analyzed in patients with RA (Figure 5). Of note, serum IL-2 concentrations showed a significant positive correlation with the TCR repertoire diversity of memory CD4+ T cells in RA (Figure 5A). This correlation was also observed when the TCR repertoire diversity was evaluated using the Renyi index with α > or < 1 (Figure 5A). In contrast, the TCR repertoire diversity of naive CD4+ T cells did not show any significant correlation with the serum concentrations of cytokines, including IL-2 (Figure 5B). Taken together, the serum IL-2 concentration also showed an association with the TCR repertoire diversity of memory CD4+ T cells in patients with RA, and the direction of the correlation differed from that of the correlation between TCR repertoire diversity and SE allele dosage.

Correlations between the TCR repertoire diversity of (A) memory and (B) naive CD4+ T cells and serum cytokine concentrations in patients with RA (n = 22). P values (−log10) were obtained by Spearman correlation analysis between the TCR repertoire diversity and serum cytokine concentrations. TCR: T cell receptor; RA: rheumatoid arthritis; IL: interleukin; TNF: tumor necrosis factor.
DISCUSSION
We demonstrated a significant and independent association of HLA-DRB1 SE alleles with the reduced TCR repertoire diversity of CD4+ T cells in patients with RA. A study demonstrated that the HLA-DR locus is a determinant of Vα gene usage as a trans-acting expression quantitative trait locus (eQTL)16. In that study, eQTL mapping was performed to determine the association between TCR V gene usage and genetic variation in a large human cohort, demonstrating that specific HLA amino acids were associated with V gene usage16. Our observation was consistent with their findings, and the significant association of the TCR repertoire diversity with HLA-DRB1, which confers the strongest genetic risk, suggested that the reduced TCR repertoire diversity plays an important role in RA pathogenesis. Our data demonstrated that decreased TCR repertoire diversity was more significant in memory CD4+ T cells than in naive T cells. There are several explanations for this observation. First, the number of HLA-DR molecules required for T cell proliferation is considerably different between naive and memory CD4+ T cells. Ten- to 100-fold greater levels of peptide-MHC complexes are required for proliferation of naive CD4+ T cells than memory T cells17. Second, some evidence suggested that the TCR repertoire of naive T cells is determined by naive T cell interaction with the self-peptide–MHC class II complex18. However, the pool of naive T cells is also formed by peptide-MHC–independent mechanisms, such as homeostatic expansion. In contrast, all memory T cells interact with the peptide-MHC class II complex during their differentiation. These differences explain why the TCR repertoire diversity of memory T cells was associated more strongly with SE alleles in RA. Abatacept (CTLA4-Ig) is clearly an effective treatment for RA, and the major target of abatacept is reported to be memory T cells in bone marrow transplant patients19. These findings suggest the importance of memory CD4+ T cells and their TCR repertoire in RA pathogenesis.
One limitation of our study is that the SE dosage-dependent decrease in the TCR repertoire diversity in HD could not be analyzed, because a homogeneous HD population regarding SE was not included in our study (Table 1). Further, the number of patients with RA was not sufficient for conducting multiple comparisons. Therefore, additional subjects are needed to analyze the TCR repertoires in patients with RA. The CD8+ T cell repertoire of patients with RA is also an important research topic that has been evaluated previously20. In our study, we focused on CD4+ T cells, rather than CD8+ T cells, because HLA-DR presents antigens to CD4+ T cells, and the association between SE and the CD4+ T cell repertoire was the primary research aim of our study. The association between the CD4+ and CD8+ T cell repertoires will be examined in the future.
Considering that SE presumably bind to certain RA-associated peptides3 and the significant association observed between SE alleles and TCR repertoire diversity in memory T cells, the skewing of memory CD4+ T cell clones driven by autoantigens presented on HLA-DRB1 SE may play a key role in the reduced TCR diversity in RA. To our surprise, TCR repertoire diversity was not associated with autoantibodies, such as anti-CCP2 antibody titers, although some citrullinated peptides bind to SE and citrullinated antigen-specific T cells were reported to be increased in patients with RA3. Our observations suggested that in addition to citrullinated antigen-specific T cells, other SE-associated autoantigen-specific T cells are expanded in RA21. In addition, alterations of other unknown SE-associated antigen-specific T cell clones could play an important role in the reduced TCR repertoire diversity in RA. Another limitation is that some patients showed lower serum anti-CCP antibody titers, even though they had an SE dosage of 2 (Table 1). This could introduce bias into the analysis. To test this hypothesis, high-throughput analysis of TCR specificity and tetramer analysis in another patient cohort are needed.
Disease activity was another significant determinant of TCR repertoire diversity. The TCR repertoire diversity of both naive and memory T cells was associated with disease activity. In RA, the levels of T cell receptor excision circle content in naive T cells are decreased and inversely correlated with disease activity22. This suggests that disease activity-related proliferation and/or selection of postthymic naive T cells are enhanced in RA patients with elevated disease activity. Because memory T cells are derived from naive T cell pools, the TCR repertoire diversity of memory T cells was also reduced in association with disease activity. Even though the precise mechanisms remain unclear, this reduced TCR repertoire diversity of naive T cells in association with disease activity was independent of SE alleles. It is not clear whether this reduction in TCR repertoire diversity causes joint inflammation or rather is a result of disease activity in RA; this is an inherent limitation of association studies. Nevertheless, the decrease in TCR repertoire diversity of memory CD4+ T cells in RA was mediated by at least 2 different processes: postthymic selection of naive CD4+ T cells and SE allele–dependent proliferation of memory CD4+ T cells.
Previous studies suggested that biased homeostatic expansion contributed to the TCR repertoire in RA5,6, and that IL-7 is a key cytokine involved in homeostatic expansion15. Interestingly, serum cytokines such as IL-2 are positively correlated with the TCR repertoire diversity of memory CD4+ T cells, but not of naive CD4+ T cells. We speculated that cytokine-induced T cell proliferation is relatively nonselective and does not contribute to the altered T cell clone distribution. Therefore, TCR repertoire diversity in RA may be regulated in both HLA-DRB1 SE allele-dependent and proliferative cytokine-dependent manners. A third limitation of our study is that the serum cytokine concentrations of the HD were not measured; additional subjects would help further elucidate these phenomena. Some patients with RA are resistant to anticytokine therapies, and regulation of T cell clones may provide an alternative therapeutic strategy for SE allele– positive patients with RA. Our previous report demonstrated characteristic gene expression profiles, especially for immune senescence-associated genes, in highly expanded CD4+T cell clones in patients with RA11. We speculate that selective deletion of memory T cells with these features is one possible mechanism for selective regulation of expanded T cell clones.
SE alleles and disease activity were negatively correlated with the TCR repertoire diversity of CD4+ T cells in RA. Our data suggest the importance of the TCR repertoire of CD4+ T cells in the pathogenesis of RA, and T cell target immunotherapy may provide a rescue therapy for patients with RA who are refractory to anticytokine therapies.
ONLINE SUPPLEMENT
Supplementary material accompanies the online version of this article.
Footnotes
Supported by the Ministry of Health, Labor, and Welfare, the Ministry of Education, Culture, Sports, Science, and Technology (KAKENHI) Grant-in-Aid for Scientific Research (C, 26461462), as well as by a Grant-in-Aid for Scientific Research (S, 23229007) from the Japan Society for the Promotion of Science.
K.Y. received financial support or fees from Astellas, BMS, MitsubishiTanabe, Pfizer, Ayumi, Takeda, Chugai, Eisai, Taisho Toyama, UCB, and ImmunoFuture. K.F. received financial support or fees from Astellas, BMS, MitsubishiTanabe, Pfizer, Ayumi, Takeda, Chugai, Eisai, Taisho Toyama, Eli Lilly, and UCB.
- Accepted for publication January 24, 2018.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}