

To the Editor:
Juvenile idiopathic arthritis [JIA; Mendelian Inheritance in Man (MIM) 604302] is the most common chronic childhood arthritis, characterized by chronic articular findings of unknown origin, with heterogeneity in disease course and systemic involvement1,2. Epidemiologic studies based on different diagnostic criteria showed varying prevalence from 0.07 to 4.01 per 1000 children across populations, an increased risk for European descendants, and different subtype distributions among ethnic groups3,4. JIA is generally known as a complex genetic trait with non-Mendelian inheritance pattern possibly resulting from interactions of multiple genetic loci and environmental factors5. However, several studies have reported causative variants in the Laccase (multicopper oxidoreductase) domain-containing 1 (LACC1; MIM 613409) gene in rare familial forms6,7,8.
We present 17 patients (10 males and 7 females) from 7 families (5 with known parental consanguinity), each with 2–4 affected members diagnosed with JIA. This study was approved by the Institutional Review Board of Istanbul Technical University (MBG.22/2014) and carried out in compliance with the Declaration Helsinki.
Clinical and laboratory findings are summarized in Table 1⇓. Mean age of patients was 15.3 ± 9.1 (range 2.8–44) years and mean disease duration 11.9 years. In 12 patients, disease onset was before age 3. Subtype of arthritis was polyarticular in 13 patients, enthesitis-related in 3 patients, and oligoarticular in 1 patient. The disease was episodic in 6 of the patients, whereas it was chronic in the others. Ten patients still had active disease and 11 were still under biologic therapy.
Demographic and clinical features of patients with novel/rare homozygous LACC1 variant.
Demographic and clinical features of patients without novel/rare homozygous LACC1 variant.
During the course of the disease, large joints were involved in all patients except B III, and in 9 of them, small joints were also involved. Joint involvement was symmetrical in 13 patients. Flexion or extension contractures developed at joints in 8 patients. No patient had gastrointestinal symptoms such as abdominal pain, vomiting, diarrhea, or oral/anal aphthous lesions that are suggestive of inflammatory bowel disease.
We first performed multipoint linkage analysis using high-density single-nucleotide polymorphism (SNP) genotyping microarrays (Illumina OmniExpress-24 BeadChip) in 2 families (A and B). A maximal cumulative LOD score of 4.81 was obtained at 13q13.3–13q14.13. Overlapping homozygosity at the linked locus was 6.8 Mb and harbored 95 genes, 38 of which were protein-coding, including the LACC1. Because of the possibility of association between rare variants in LACC1 and familial form of JIA, we prioritized LACC1 for sequencing. In both families we found novel/rare, pathogenic/likely pathogenic variants segregating with the disease by Sanger sequencing of the coding regions and exon-intron junctions of LACC1. Novel variant c.3G>A (p.0) in family A is deduced to disrupt the translational initiation codon (Figure 1), and Western analysis showed absence of protein production (data not shown). Rare variant c.1240C>T [p.(Arg414Ter); rs184370809] in family B resulted in premature termination at codon 414, causing the deletion of the terminal 17 amino acids. Our findings verified that LACC1 gene defects underlie the familial form of juvenile arthritis and prompted us to sequence the coding regions of LACC1 in the remaining 5 families. Some other rare variants were detected in 2 families. Homozygous variant c.988_990del [p.(Ile330del); rs776489319] in family C is an in-frame deletion that resulted in the absence of the isoleucine residue at position 330. Patients of family F carried a common homozygous variant c.760A>G [p.(Ile254Val); rs3764147] together with novel heterozygous c.1109G>A [p.(Cys370Tyr)]; however, an unaffected brother (F III) also had the same genotype, indicating that the 2 variants are not sufficient to cause juvenile arthritis on their own but could be susceptibility alleles. Patients in the remaining 3 families (D, E, G) did not carry any rare/novel variants in LACC1.
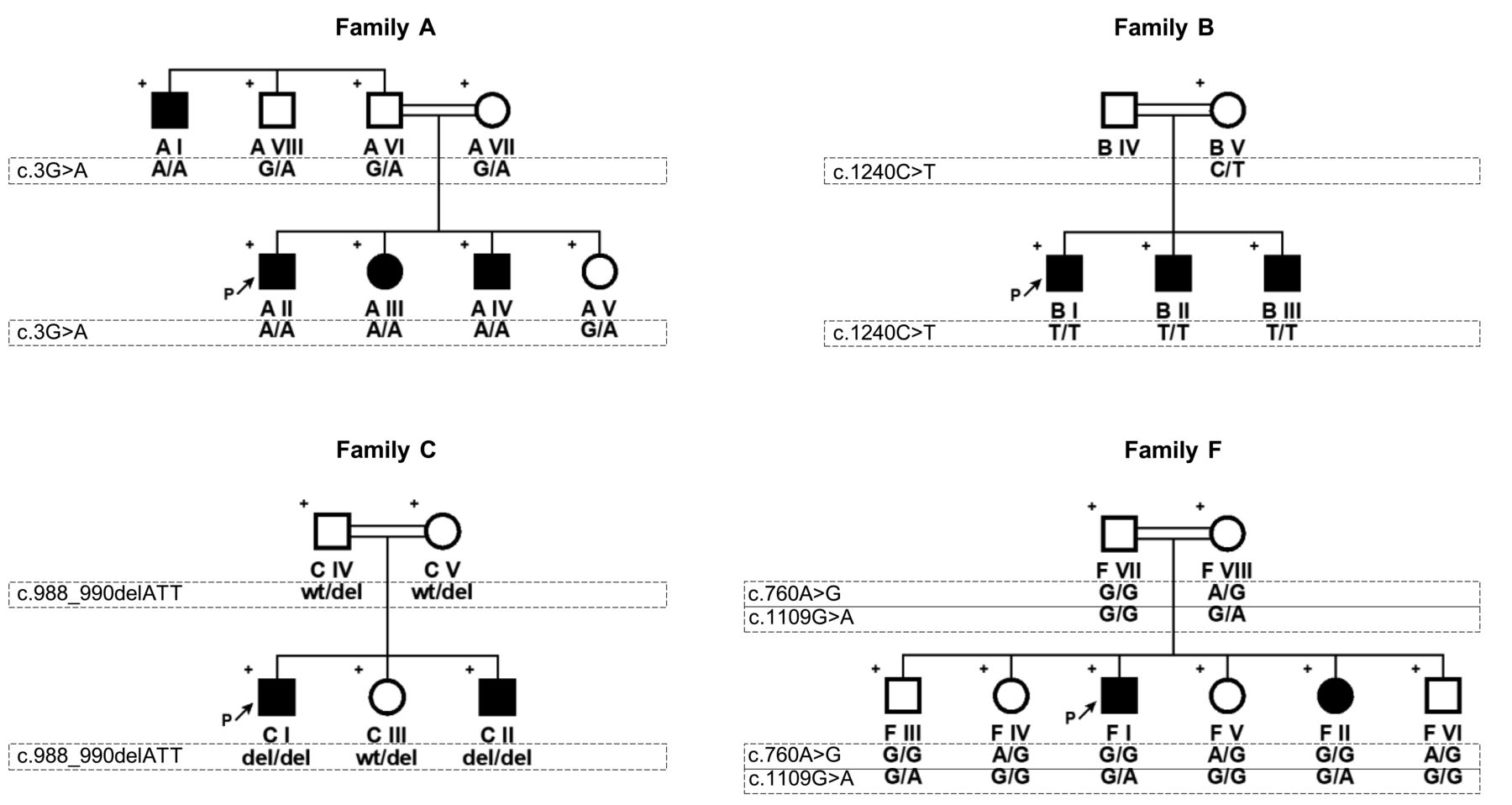
Pedigrees of the families. Plus sign indicates individuals included in the genetic analysis. Arrows show index patients. Variants are defined according to NM_001128303.1. Wt: wild type; del: deletion.
Further, the 2 patients in family E, which was the only consanguineous family without a possible causative LACC1 variant, were also subjected to SNP genotyping to investigate whether they shared genotypes in the LACC1 region. SNP genotypes in the LACC1 region were different, excluding shared compound heterozygosity and indicating that LACC1 was not the causative gene in this family.
Our findings confirm that LACC1 variants can be responsible for the recessive form of juvenile arthritis. Age of onset appears to be earlier in patients with LACC1 pathogenic/likely pathogenic variants. The finding that affected siblings in family E do not share genotypes indicates genetic heterogeneity in familial juvenile arthritis for the first time, to our knowledge, assuming Mendelian inheritance. Future genetic studies to assess the contribution to JIA of variants in LACC1 could clarify whether LACC1 variants underlie early onset JIA as in all reported cases. Genome-wide studies are essential to understand the contribution of genetic factors to the disease.
Our findings verify that LACC1 pathogenic variants can cause familial juvenile arthritis. Because juvenile arthritis has high clinical variability, as observed here even within the same family, we propose that screening patients with JIA, especially those with very early onset, for LACC1 variants can be beneficial to patients and families.
Footnotes
This study was supported by the Technological and Scientific Research Council of Turkey (Grant 114Z829).








{kind=link}