

Abstract
Objective. Inflammatory ocular disease (IOD) is a rare but severe extraarticular manifestation of rheumatoid arthritis (ExRA) with high mortality. The aim of our study was to examine clinical characteristics of IOD in rheumatoid arthritis (RA) and their effect on disease severity and outcomes in recent years.
Methods. A retrospective cohort of RA patients with IOD evaluated between 1996 and 2013 was assembled and compared to RA comparators without IOD and matched for age, sex, and disease duration.
Results. We identified 92 patients (69% female; mean age 62 yrs) with IOD: 33 scleritis, 23 episcleritis, 21 peripheral ulcerative keratitis (PUK), 14 uveitis, and 1 with orbital inflammation. The majority of patients with scleritis, episcleritis, and PUK was seropositive versus uveitis (> 80% vs 62%, p = 0.048). PUK and scleritis were more symptomatic compared to episcleritis and uveitis, and often required systemic therapy. Time to resolution was longer in scleritis than episcleritis (p = 0.01). PUK, scleritis, and uveitis had severe ocular sequelae. Prevalence of severe ExRA (18% vs 4%, p = 0.004) and dry eye syndrome (42% vs 26%, p = 0.024) was higher among patients with IOD than comparators. The incidence of new ExRA over 5-year followup was also higher among cases (29% vs 11%, p = 0.022). Ten-year survival was similar among RA patients with and without IOD (66% vs 64%, p = 0.56), with no differences noted among IOD subtypes.
Conclusion. This large single-center study highlights the variable presentation and outcomes of IOD in RA. Although ocular complications are associated with significant morbidity, it is reassuring that survival among those with IOD is now similar to those without ocular disease.
Ocular inflammation can develop in many systemic autoimmune rheumatic diseases, and if untreated can lead to severe, irreversible vision loss. Rheumatoid arthritis (RA) can be complicated by extraarticular manifestations (ExRA)1 in up to 40% of cases2. Eye involvement is common, and reported in 28%2 to 39%3 of patients, depending on the study. It may rarely be the presenting sign of disease. Despite significant improvements in RA treatment in the last few decades, the incidence of ExRA remains largely unchanged other than a decline in rheumatoid vasculitis and more frequent dry eye syndrome4.
Ophthalmological manifestations of RA can be diverse and present as surface disease or inflammatory ocular disease (IOD)5 affecting the episclera, sclera, cornea, uvea, retina, or orbit. The former, clinically presenting as dry eye syndrome or keratoconjunctivitis sicca (KCS), can affect up to 28% of patients with RA3. RA accounts for a sixth of all patients with scleritis6. Peripheral ulcerative keratitis (PUK) can be complicated by rapid corneal keratolysis (corneal melt syndrome), perforation of the globe, and visual failure. The onset of necrotizing scleritis (NS) and PUK may precede or be concurrent with systemic rheumatoid vasculitis (SRV)7. Up to 40% of patients with SRV die within 5 years because of damage from vasculitis and/or consequences of immunosuppressive therapy8,9,10,11. Reports from the prebiologic era note high mortality from cardiovascular (CV) events among patients with NS and PUK7.
Risk factors for IOD are not well reported in the literature. Ocular complications such as cataracts and glaucoma can result from longterm glucocorticoid (GC) use. Rarely, scleritis, uveitis, and orbital myositis can result from use of bisphosphonates administered to manage osteoporosis in patients with RA12.
In the absence of evidence-based recommendations for management and poor prognosis, IOD in RA is often treated with intense immunosuppression such as high-dose GC and cyclophosphamide (CYC). The role of treat-to-target strategies and use of disease-modifying antirheumatic drugs (DMARD) and biologics in refractory IOD is unclear, but benefit of the latter has been demonstrated13.
The aim of our study was to characterize the clinical correlates of IOD in a cohort of patients with RA evaluated between 1996 and 2013.
MATERIALS AND METHODS
Study design
In this retrospective cohort study, patients over age 18 years evaluated at the Mayo Clinic, Rochester, Minnesota, USA, between January 1, 1996, and December 31, 2013, with an International Classification of Diseases, 9th revision (ICD-9) code for RA were identified. From these, subjects with an IOD diagnosed at any point after (or within 6 mos prior) RA diagnosis were included. IOD studied included episcleritis/scleritis (ICD-9 379.0), PUK (ICD-9 370), uveitis (ICD-9 364), retinal vasculitis (362.18), and nonspecific orbital inflammation (ICD-9 376.11). The complete inpatient and outpatient medical records were reviewed by 1 author (CSC) to confirm that patients met the 1987 American College of Rheumatology classification criteria for RA and that IOD diagnosis was confirmed by an ophthalmologist. Patients were followed from diagnosis until last followup visit or death. Patients with IOD diagnosed 6 months prior to RA diagnosis were excluded from the analysis. Only records of patients who did not deny use of their medical records for research purposes were reviewed, per Minnesota law. The study was approved by the Mayo Clinic Institutional Review Board (IRB 14-006237).
Data collection
Data abstracted from medical records were collected on a standardized data collection form using the Research Electronic Data Capture tool hosted at the Mayo Clinic14. For all subjects, data abstracted were demographics, smoking status, clinical features of RA (articular and ExRA)15,16, laboratory studies, medications, and comorbidities. RA disease activity was recorded at onset of IOD (index date), and defined either objectively by the 28-joint Disease Activity Score (< 3.2 low disease activity vs > 3.2 moderate to high disease activity) or subjectively based on physician judgment (Active: RA requiring therapeutic intervention; Inactive: no change in therapy). ExRA were classified as severe as previously defined by the Malmo criteria (Table 1)16.
Clinical characteristics of rheumatoid arthritis among patients with inflammatory ocular disease seen at the Mayo Clinic, Rochester, Minnesota, USA, between 1996 and 2013. Values are median (25th percentile, 75th percentile) or n (%). Percentages are calculated among patients with available data.
Cases (RA patient with IOD)
The first and all subsequent episodes of IOD for each case were recorded. Scleritis and episcleritis were classified according to Watson and Hayreh17 and uveitis as anterior or posterior18, where possible. Ocular history included cataracts, glaucoma, epiretinal membrane, corneal scar, surgery or eye infections in the previous 4 weeks, hydroxychloroquine retinopathy, and contact lens use. Episodes of IOD in which underlying etiology was determined to be other than RA were excluded. Visual acuity (VA) was expressed as logarithm of the minimal angle of resolution (LogMAR) and recorded prior to and at IOD onset, posttreatment, and at last followup. The ocular outcomes evaluated included resolution (based on ophthalmology examination), remission (defined as no IOD activity for at least 6 mos), relapse, vision loss (defined as loss of at least 2 Snellen lines), and complications (including cataract, ocular hypertension, central serous chorioretinopathy, corneal scar or thinning, scleral thinning, cystoid macular edema, and exudative retinal detachment). The incidence of new ExRA and CV events in the 5 years following IOD onset was noted.
Comparator subjects (RA patients without IOD)
For each case, an RA patient without IOD was randomly selected from among patients seen in the time frame of interest (1996–2013) and matched for age, sex, calendar year, and disease duration. The index date for comparator subjects was defined as the visit date closest to the first IOD diagnosis date in the matched case.
Statistical analysis
Descriptive statistics were used to summarize the characteristics of patients with and without IOD. Chi-square and rank sum tests were used to compare characteristics between groups. For patients with more than 1 IOD (or bilateral eye involvement) at presentation, a single IOD type was chosen for comparisons (with preference for scleritis over other types of IOD, or by random selection when both IOD were the same type).
Kaplan-Meier methods were used to estimate survival, resolution, and remission rates over time. Comparisons of survival rates between patients with and without IOD were performed using log-rank tests. Comparisons adjusted for age differences between the groups were performed using Cox models.
All episodes of IOD were included in the analyses of VA and ocular complications. Mixed models including random effects to account for multiple episodes of IOD in the same patient were used to compare VA between IOD types. Frailty models (an adaptation of Cox models with random effects per patient) were used to compare time to complication between patients with different types of IOD. Statistical analyses were performed using SAS Version 9.4 (SAS Institute) and R 3.1.1 (R Foundation for Statistical Computing).
RESULTS
Patient characteristics
A total of 92 patients with RA (69% female; mean age 62 yrs) were identified with IOD in 1996–2013 including 33 (44 eyes) with scleritis, 23 (30 eyes) episcleritis, 21 (28 eyes) PUK, 14 (16 eyes) uveitis, and 1 (1 eye) with orbital inflammation. Median followup was 4.1 years [interquartile range (IQR): 1.5–9.4 yrs].
Patients with episcleritis were younger while those with uveitis were relatively older (53.5 yrs vs 69.3 yrs, p = 0.032; Table 1). Joint erosions were as common in patients with uveitis as in scleritis and more common among patients with PUK (p = 0.51). More than 80% of patients with scleritis, episcleritis, and PUK were seropositive, versus 62% with uveitis (p = 0.048). Disease activity of RA at IOD onset seemed absent or mild in the majority of patients.
Severe ExRA before IOD onset were more common in patients with scleritis and episcleritis than among those with PUK and uveitis. More patients with PUK had Sjögren syndrome (20%, p = 0.10). Only 8% of patients with uveitis had rheumatoid nodules compared to 35% and 36% of patients with PUK and scleritis, respectively (p = 0.19). Radiographic erosions were present in 63% of patients. Around 70% of patients were taking DMARD at IOD onset. Patients with uveitis and PUK were somewhat less likely to be taking GC therapy, and the latter were also less likely to be taking biologics at IOD onset.
The prevalence of CV diseases and their risk factors (coronary artery disease, ischemic heart disease, transient ischemic attack, stroke, diabetes, and hyperlipidemia) were relatively higher among patients with uveitis, but did not reach statistical significance (Supplementary Table 1, available from the authors on request).
Clinical presentation of IOD
Table 2 summarizes characteristics at presentation of IOD. Scleritis was anterior in 31/33 (94%; 2 diffuse, 6 nodular, and 3 necrotizing). Two patients had scleromalacia perforans. All uveitis cases were anterior in distribution.
Clinical correlates of inflammatory ocular disease at presentation among patients with RA seen at the Mayo Clinic, Rochester, Minnesota, USA, between 1996 and 2013. Values are median (25th percentile, 75th percentile) or n (%).
Uveitis was less often bilateral compared to other IOD (14% uveitis vs 30% episcleritis, 33% PUK, and 33% scleritis). Patients with episcleritis, scleritis, and uveitis almost always presented with eye redness, while redness was present only in 65% of patients with PUK (p = 0.004). Ocular pain and headache were more common in scleritis compared to episcleritis. In general, episcleritis presented less often with pain or vision alteration. Vision loss or dry eye/foreign body sensation was especially common in PUK.
Eye surgery within the previous 4 weeks was reported by 2/14 uveitis patients and 1 scleritis patient (out of 33) who also had concomitant uveitis. One patient with uveitis also had an eye infection in the 4 weeks prior to presentation. Patients with uveitis also had a higher prevalence of cataract prior to onset (57% in uveitis vs 26% in episcleritis, 38% in PUK, and 33% in scleritis) that was likely related to their older age. All 3 patients with a history of contact lens use at IOD onset had episcleritis.
Laboratory studies were frequently normal at presentation of IOD, particularly blood counts and inflammation markers. Antineutrophil cytoplasmic antibodies (ANCA) were positive in 6 patients (perinuclear ANCA in all 6), 2 of whom had severe ExRA (1 neuropathy and 1 pericarditis); 3 had rheumatoid nodules and 4 KCS.
Treatment of IOD episode
Ocular and systemic treatment for IOD is outlined in Table 3. Patients with episcleritis and uveitis were more often treated with topical agents. Scleritis and PUK often necessitated the use of systemic GC, CYC, additional DMARD, or biologic therapy. Methotrexate was the most frequently used DMARD. Antitumor necrosis factor agents were preferred when a biologic was initiated for treatment. Surgical treatment was necessary in 7 patients (14 procedures), all with PUK.
Treatment strategies used for inflammatory ocular disease in rheumatoid arthritis patients seen at the Mayo Clinic, Rochester, Minnesota, USA, 1996–2013. Values are n (%).
Outcomes
Figure 1 illustrates the time to resolution and remission for first episode of each IOD subtype. The cumulative incidence of remission (± SE) for scleritis at 1 year was only 40 ± 9%, as compared to 60 ± 11% for episcleritis, 73 ± 10% for PUK, and 83 ± 11% for uveitis. No difference was identified in time to relapse between the various IOD subtypes (4 group log-rank p = 0.85; HR compared to episcleritis for scleritis: 1.52, 95% CI 0.36–6.42; PUK: 1.30, 95% CI 0.26–6.48; uveitis: 0.65, 95% CI 0.07–6.28). However, patients with scleritis demonstrated a trend toward higher risk of developing a second IOD subtype during followup (4 group log-rank p = 0.038; HR compared to episcleritis for scleritis: 3.56, 95% CI 0.96–13.21; PUK: 0.41, 95% CI 0.04–3.98; uveitis: 1.84, 95% CI 0.37–9.14).
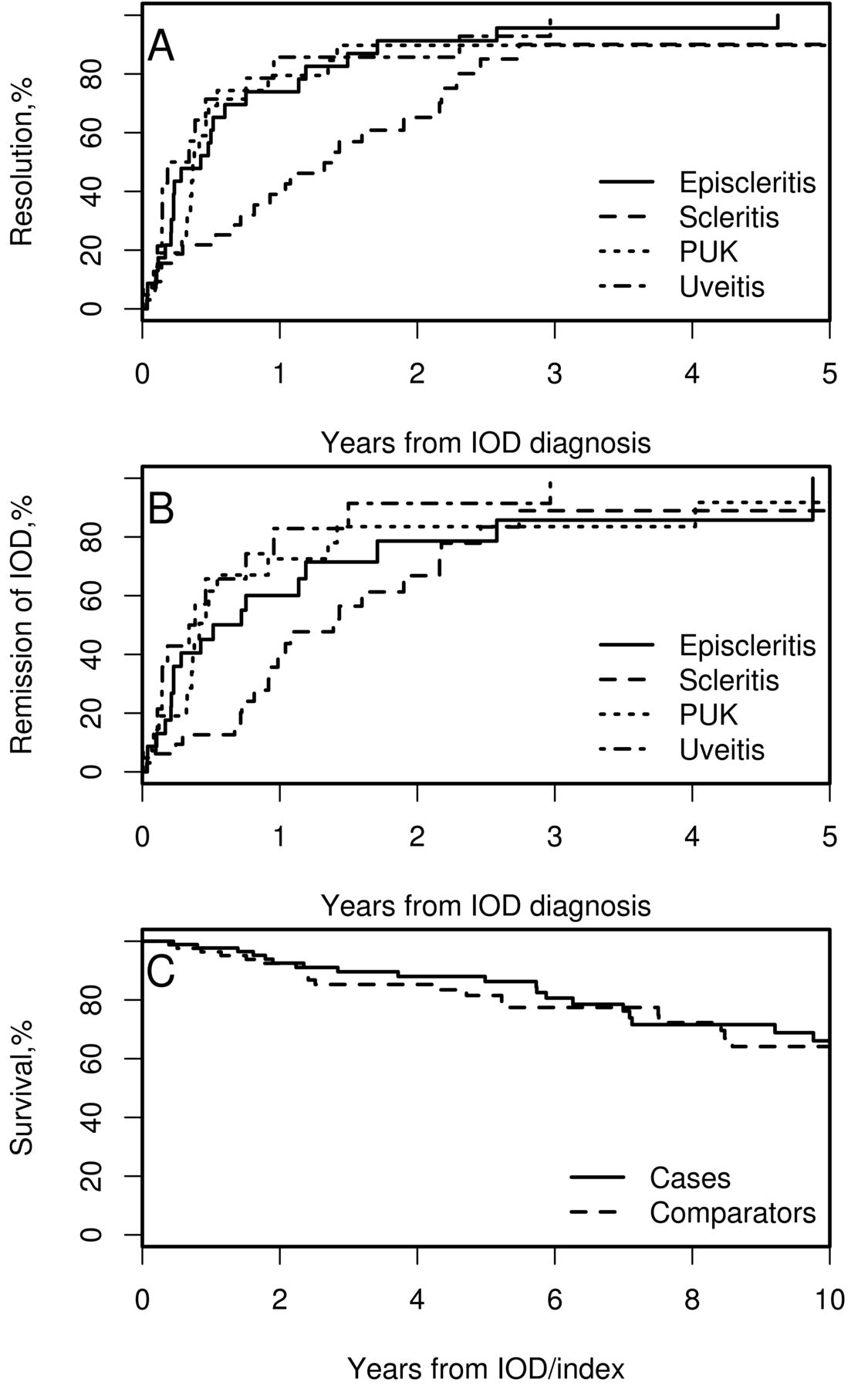
A. Cumulative incidence of resolution of the first episode of IOD in patients with episcleritis, scleritis, PUK, and uveitis. B. Cumulative incidence of remission of the first episode of IOD in patients with episcleritis, scleritis, PUK, and uveitis. C. Survival rates in patients with IOD (cases) and patients without IOD (comparators). IOD: inflammatory ocular disease; PUK: peripheral ulcerative keratitis.
Table 4 summarizes ocular outcomes and complications of IOD. Affected eyes of patients with episcleritis had the most preserved VA at each point during evaluation, in comparison to other IOD, all of whom demonstrated significant decline in vision compared to their best VA (p = 0.001).
Ocular outcomes and complications of episodes of inflammatory ocular disease in patients with rheumatoid arthritis seen at Mayo Clinic, Rochester, Minnesota, 1996–2013.
Cataract development was significantly higher in patients with PUK and scleritis, who were also treated most often with systemic GC compared to other IOD. Intraocular hypertension and cystoid macular edema were more common in scleritis and uveitis, even after accounting for topical and systemic GC therapy. Scleral thinning was highest in scleritis, and corneal scars occurred only in PUK.
Figure 1C shows that survival at 10 years was comparable at 66% ± 7% in cases and 64% ± 7% in comparators (p = 0.56). Results were similar in subgroup analyses of each IOD type compared to its own comparators. Ten-year survival rate was not different between various IOD, after correcting for age (p = 0.80). At 5 years of followup, only 9% of patients with uveitis had a new ExRA as compared to 39% of episcleritis, 38% of PUK, and 29% of scleritis (p = 0.34).
No differences were found among different IOD for CV events during 5 years of followup, although a higher prevalence was noted in patients with PUK (31% compared to 17% in episcleritis, 12% in scleritis, and 18% in uveitis; p = 0.61).
Risk factors and outcomes for IOD versus comparators
Ninety IOD cases were matched to 90 control patients to identify risk factors for IOD in RA (Table 5). For 2 patients, controls meeting all eligibility criteria could not be identified and these were excluded from the analysis. The median disease duration was 10.2 years versus 9.9 years (p = 0.61) and median followup 5.7 years (IQR 1.9–10.2) for patients with IOD and 4.4 years (IQR 2.0–9.0) for comparators. Joint disease severity was similar between the 2 groups while ExRA were more frequent in patients with IOD. In particular, neuropathy and severe ExRA were more frequent in cases (7% vs 0%, p = 0.012 and 18% vs 4%, p = 0.004, respectively). KCS was significantly more frequent among patients with IOD (42% vs 26%, p = 0.024). No differences were noted in RA treatment between cases and controls.
Comparison of clinical characteristics among patients with rheumatoid arthritis who have inflammatory ocular disease (IOD) and their comparators without IOD matched for age, sex, and disease duration seen at Mayo Clinic, Rochester, Minnesota, USA, between 1996 and 2013. Values are median (25th percentile, 75th percentile) or n (%). Percentages are calculated among patients with available data.
Cases were more commonly receiving treatment with bisphosphonates than comparators (25% vs 10%, p = 0.013). There was no significant difference in the prevalence of rheumatoid factor, cyclic citrullinated peptide, antinuclear antibody, or ANCA between cases and controls.
The incidence of new ExRA in the 5 years of followup was higher in patients with IOD (29% vs 11%, p = 0.022). Most of these were new or worsening rheumatoid nodulosis. Four patients with IOD and 1 comparator developed cutaneous vasculitis.
The prevalence of CV comorbidities was similar between cases and controls (16% vs 21%, respectively, p = 0.34). The incidence of new CV events at 5 years of followup was no different between the 2 groups (p = 0.47).
IOD incidence prior to onset of RA
Among patients who developed IOD more than 6 months prior to their diagnosis of RA (Supplementary Table 2, available from the authors on request), the median time between IOD onset and RA diagnosis was 21.8 months (IQR 13.2–41.4 mos). Three of 6 patients were seropositive. None of the 5 patients with a complete ophthalmologic history reported KCS before IOD onset. Interestingly, the only patient with a history of eye surgery within 4 weeks from IOD onset developed uveitis. During the followup period (median 89.6 mos, IQR 70.4–105.8), at least 1 relapse of IOD occurred in half of patients, all post-RA onset.
DISCUSSION
Our study describes the largest single-center series of IOD in patients with RA in the recent era of biologic use and is the first, to our knowledge, to describe clinical correlates of the various IOD in these patients. Despite the advent and extensive use of biologics for RA, IOD remains an ExRA that is still largely managed with alkylating agents, conventional DMARD, and high-dose GC.
Most of the literature on IOD in RA is from the prebiologic era, and includes case series of scleritis and PUK7,19,20,21,22,23. Visual and systemic outcomes were often poor and mortality alarmingly high at 30–50% over 5 years7,19,20. Even in a recent report24, 11 of 38 patients with rheumatoid keratolysis died after brief intervals despite intervention. In general, patients treated with aggressive immunosuppression fare better7. Only small numbers of non-infectious IOD in RA have been reported25,26. Uveitis and episcleritis17,19,27,28,29 are well described, but data on patients with RA are limited.
In our study, patients with scleritis generally had longstanding, seropositive, and erosive RA, and frequently developed new ExRA within 5 years of IOD. Patients with episcleritis were significantly younger and had a similar and quite high prevalence of ExRA before IOD onset and during followup. A majority of patients with PUK had seropositive erosive RA, but with minimal to no disease activity prior to IOD onset. They had a higher prevalence of Sjögren syndrome, and developed frequent ExRA and CV events during followup. Patients with scleritis and PUK experienced significantly more vision loss and ocular complications in comparison to episcleritis and uveitis. More than one-third of patients with PUK required surgical intervention to preserve ocular integrity. Uveitis was more prevalent among elderly onset RA and occurred earlier in the course of disease than other IOD. Development of ExRA during followup of uveitis was less frequent and visual outcome was good, except for a higher incidence of glaucoma.
Histopathologic studies have showed that both scleritis and PUK are often characterized by a vasculitic process occurring in the eye21,30. Seropositivity is a risk factor for rheumatoid vasculitis9,31 and ExRA, so the high prevalence of seropositive patients in our current study is not surprising. The high incidence of ExRA following scleritis has also been reported previously20. New ExRA were more frequent, especially after PUK onset (38% as compared to 9% of uveitis and 29% of scleritis), likely reflecting shared disease mechanisms including genetic predisposition32, which also explains clustering of severe ExRA in community-based samples of patients with RA33. Patients with uveitis were more frequently seronegative (8/13) and had less frequent ExRA. Two patients developed uveitis within 4 weeks from a previous ocular surgery and 1 had a preceding eye infection.
KCS was significantly higher in patients with IOD than in comparators (episcleritis 43%, scleritis 46%, and PUK 45%). The prevalence of KCS in PUK was lower in our study than reported previously (71%)24. The prevalence of ocular surface diseases ranged between 61.9% and 100% in other descriptions of PUK22,34. KCS has been reported in 14.2% of patients with episcleritis and 12.9% of patients with scleritis19.
In the current study, bisphosphonate use was significantly higher among patients with IOD than comparators. While this relationship has been reported by others, it is possible that confounding by indication could explain some of the association35,36,37 because these patients are more likely to have exposure to high-dose GC and thus higher risk of osteopenia, osteoporosis, and fractures, requiring prophylaxis or treatment with bisphosphonates.
The most common complication of IOD was cataract, mainly secondary to GC treatment. Ocular hypertension developed in 17% of patients with scleritis, similar to an earlier report19. Scleral thinning occurred in 51% of patients with scleritis despite treatment, similar to 5/9 in untreated patients from an earlier report7. Corneal thinning occurred in 57% of patients with PUK in the current study; there are no systematic reports about this in the literature. Corneal graft was performed less frequently in our cohort as compared to a previous report (2/21 vs 5/9)22. Loss of VA was worst among patients with PUK and least among patients with episcleritis.
Survival in patients with IOD was similar to comparators, and for scleritis, much improved over previous reports. Mortality of over 36% over 3 years in patients with RA-associated scleritis (5 of 14) has been reported19,20. In patients with scleritis untreated by immunosuppressive agents, a mortality of 66.7% over 2 years has been reported, and 7.7% in patients treated with CYC (4.1 yrs followup)7. The advancements in RA management, including aggressive use of DMARD and biologic treatments and treat-to-target strategies, have reduced the occurrence of vasculitis and overall mortality due to ExRA over the years4. This trend is reflected in the lower rates of vasculitis and CV disease in patients with scleritis in the current study compared to earlier reports19,20. CV disease occurred in 15% of our patients compared to 60% in previous reports19; differences in CV case ascertainment, management, and followup may account for some of these differences.
Of 2 reports of mortality among patients with RA-associated PUK before 1996, 1 found that all 5 patients treated with immunosuppressive drugs survived during a followup of 5.8 years7, while 37.5% of 8 patients not treated with immunosuppressive drugs died during 12.0 years of followup. Palay, et al reported a mortality of 48% over 5 years of followup in a cohort undergoing keratoplasty for corneal melt23. More recently, 10-year mortality of 25% has been reported in RA patients with keratolysis diagnosed between 1987 and 200224. Mean disease duration was 15 years, but RA severity or comorbidities were not reported.
Unlike previous authors17,19,23, we did not find any differences in the incidence of CV events in scleritis, episcleritis, or uveitis cases versus comparators. Although not reaching statistical significance, in the current study, patients with PUK had a 3-fold higher incidence of CV events in comparison to controls.
Limitations of our study are largely related to its retrospective design and the potential for referral bias. However, most of the information collected comes from detailed rheumatology and ophthalmology visits, so clinically important events are less likely to be missing. The risk of misclassification of IOD was reduced by review of doubtful cases by a rheumatologist (AM) and an ophthalmologist (WMS).
Our study highlights the clinical presentation, treatments, and outcomes of IOD in RA. The systemic character of the disease is reflected in the association of PUK and scleritis, with increased risk for developing new ExRA. Uveitis tends to occur in patients with seronegative RA and has a lower risk for ExRA. KCS is more common among patients with IOD, but whether optimal management of this symptom might reduce risk for major eye disease remains unclear. Reassuringly, there has been a remarkable improvement in survival in patients with IOD compared to earlier reports in the literature, with overall survival that is now comparable to that of RA patients without IOD.
Footnotes
Work funded by the Mayo Clinic Margaret Harvey Schering Clinician Career Development Award Fund for Arthritis Research and supported by Grant Number UL1 TR000135 from the US National Center for Advancing Translational Sciences.
- Accepted for publication November 9, 2017.








{kind=link}