

Abstract
Objective. Anticitrullinated protein antibodies (ACPA) have major diagnostic significance in rheumatoid arthritis (RA). ACPA are directed against different citrullinated antigens, including filaggrin, fibrinogen, vimentin, and collagen. The presence of ACPA is associated with joint damage and extraarticular manifestations, suggesting that ACPA may have a significant role in the pathogenesis of RA.
Methods. To verify the effect of ACPA on RA-immune cells, peripheral blood mononuclear cells (PBMC) from cyclic citrullinated peptide (CCP)–positive patients with RA and healthy controls were cocultured in vitro with ACPA. ACPA-positive stained cells were analyzed by flow cytometry and the effect of ACPA on mRNA expression levels was evaluated by real-time PCR. We tested whether the stimulatory effects induced by ACPA could be inhibited by the addition of a new multiepitope citrullinated peptide (Cit-ME).
Results. We found that ACPA bind specifically to PBMC from CCP-positive patients with RA through the Fab portion. ACPA induce upregulation of pathogenic cytokine expression (4- to 13-fold increase) in PBMC derived from CCP-positive patients with RA. Moreover, ACPA upregulated IL-1β and IL-6 mRNA expression levels by 10- and 6-fold, respectively, compared to control IgG. Cit-ME, a genuine ligand of ACPA, inhibited the ACPA-induced upregulation of IL-1β and IL-6 by 30%.
Conclusion. ACPA bind to a limited percentage of PBMC and upregulate inflammatory cytokine expression, suggesting that ACPA is involved in RA pathogenesis. Targeting ACPA to decrease their pathogenic effects might provide a novel direction in developing therapeutic strategies for RA.
Rheumatoid arthritis (RA) is an autoimmune disease characterized by progressive synovial inflammation and joint destruction. Many of the RA-specific autoantibodies are generated against citrullinated antigens and are known as anticitrullinated protein antibodies (ACPA)1. ACPA can be detected in almost 80% of patients with RA2 and appear years prior to the onset of symptoms. Moreover, its presence was shown to correlate with joint destruction in RA3,4. Presence of ACPA has been linked to risk factors for RA, most notably with the HLA-DRB1 shared epitope alleles and cigarette smoking5,6,7,8,9,10. ACPA is used as a diagnostic marker, detected with the commercial anticyclic citrullinated peptide (anti-CCP) assay. It was reported to be more specific than rheumatoid factor for diagnosing RA11,12, and was included in the new American College of Rheumatology (ACR)/ European League Against Rheumatism RA classification criteria13. The major targets for ACPA are citrullinated proteins (i.e., proteins that underwent posttranslation conversion of arginine to citrulline residues). In RA, many proteins such as filaggrin, fibrinogen, vimentin, and collagen type II are subject to citrullination14. However, even if the antigenic ligands of ACPA are known, the mechanisms of their involvement in disease mediation remain obscure. Although ACPA itself was not shown to induce arthritis in animal models, its pathogenic effect was demonstrated in experimental murine models of arthritis15,16. ACPA injected in mice enhances osteoclast activation, leading to bone loss through excessive IL-8 production17. ACPA was found to induce painlike behavior in mice by activating sensory neurons by upregulation of IL-8 released from osteoclasts18. These findings are also supported by in vitro demonstration of the ability of citrullinated vimentin autoantibodies to bind osteoclastic linage cells and convert their differentiation into bone resorbing cells, promoting bone loss19. ACPA-containing immune complexes activate tumor necrosis factor–α (TNF–α) secretion from macrophages of patients with RA by binding to Fcγ receptors20,21. Macrophage colony-stimulating factor (M-CSF)–differentiated macrophages are abundant in the synovium of patients with RA22. ACPA induce proinflammatory cytokine response in M-CSF-differentiated macrophages and contribute to synovitis23,24. In addition, ACPA enhance extracellular signal-regulated kinase 1/2– and c-Jun N-terminal kinase–signaling that leads to activation of nuclear factor (NF)–κB and production of TNF–α in monocytes/macrophages by binding to membrane-citrullinated 78 kDa glucose-regulated protein (Grp78)25,26. Neutrophils are targeted by ACPA, through RA synovium being enriched with infiltrating neutrophils that are activated through the process of neutrophil extracellular trap (NET). ACPA seem to contribute to NETting of neutrophils in synovial tissues of patients with RA27. ACPA also appear to directly modulate platelet activation because ACPA titers are strongly correlated with markers of platelet activation in RA28.
ACPA circulate in the blood years prior to arthritis clinical signs and might affect peripheral blood cells that participate in the RA disease course. Study aims were to determine the following: (1) how and to what extent ACPA bind to peripheral blood cells of CCP-positive patients with RA; and (2) whether a consequence of its direct binding could be a change in the mRNA expression profile. In addition, we also tested whether a multiepitope citrullinated peptide (Cit-ME) can modify the effects of ACPA on cytokine gene expression.
MATERIALS AND METHODS
Human subjects and sample procurement
A total of 32 CCP-positive patients with RA and 27 healthy controls were included in the study. All patients fulfilled the 2010 ACR criteria for RA. The study protocol was approved by the Medical Ethics Review Board of Sheba Medical Center, Tel Hashomer, Israel (Sheba Helsinki approval number 9247-12-SMC). All participants signed an informed consent form prior to the initiation of the study.
Detection of ACPA
Sera from patients were tested for ACPA using the commercial ELISA QUANTA Lite CCP3 IgG kit (Inova Diagnostics), according to the manufacturer’s instructions. Sera with results < 25 U/ml were defined as negative and sera with results ≥ 25 U/ml, as positive.
ACPA affinity purification
ACPA were affinity purified using Hitrap NHS-activated (GE Healthcare Bio-Sciences Corp.) column chromatography according to the manufacturer’s instructions. Briefly, a mixture containing 1 mg of each of the following citrullinated peptides was coupled to 1 ml column: Cit-fillagrin 306-SHQEST(Cit)G(Cit)SRGRSGRSGS-324-K-NH229; Cit-collagen 359-A(Cit)GLTG(Cit)PGDA-369-K-NH230; Cit-vimentin 65-SAVRA(Cit)SSVPGVR-77-K-NH231; and Cit-β-fibrinogen 60-(Cit)PAPPPISGGGY(Cit)A(Cit)-74-K-NH232. The Cit-collagen, Cit-filaggrin, and Cit-vimentin peptides were synthesized manually by solid-phase peptide synthesis as previously described33 by Fruzsina Babos and Anna Magyar at the Research Group of Peptide Chemistry, Hungarian Academy of Sciences. The Cit-β-fibrinogen peptide was obtained from GL Biochem.
A pool of 5 ml of sera from 8 different RA patients with high ACPA titers (> 180) and who were positive for each of the citrullinated peptides that were linked to the column was diluted 1:1 in binding buffer (20 mM sodium phosphate buffer, pH 7.4) and run through the column for 48 h. ACPA was eluted from the column using 0.2 M glycine-HCl pH 2.2, neutralized to pH 7, and dialyzed against phosphate buffered saline (PBS). The eluted antibodies were concentrated with Millipore Amicon Ultra-15, 50K, centrifugal microconcentrators (Amicon Corp.), and were used for in vitro assays. The concentration of the ACPA was determined by QPRO-BCA standard kit (Cyanagen Srl), and its specificity was determined by positivity of all serial dilutions (1:100–1:50,000) in CCP3 IgG ELISA test. All antibodies were found to be negative for bacterial endotoxin Limulus Amebocyte Lysate test (Pierce) at the concentrations used.
In vitro experiments
Peripheral blood mononuclear cells (PBMC) were isolated from heparinized venous blood using lymphoprep (Amersham Biosciences Europe GmbH). Cells were cultured in RPMI 1640 medium containing 10% bovine fetal serum supplemented with penicillin (100 U/ml), streptomycin (100 g/ml), 2 mmol/l L-glutamine, and 50 µM 2-β-mercaptoethanol. The cells were incubated for 24 h at 37°C with the experimental antibodies.
The PBMC were exposed to 400 ng/ml of the experimental antibodies ACPA, human IgG (ChromPure Human IgG, whole molecule; Jackson Immunoresearch Inc.) or human anti-DNA monoclonal autoantibody that expresses the 16/6 Idiotype (16/6 Id)34. All experiments were performed with the affinity purified ACPA whereas in Figure 1 additional commercial ACPA was used, polyclonal antibody, generated against citrullinated filaggrin peptide (rabbit anti-CCP; Bioss-USA).

ACPA modulates IL-1β and IL-6 mRNA. PBMC were cocultured in the presence of ACPA or IgG for 24 h, then RNA was extracted. (A) Real-time PCR quantification of IL-1β and IL-6 expression in the upper panel was calculated compared to IgG (considered as 1; CCP-positive RA, n = 15 and healthy control, n = 10). (B) Relative expression was calculated following incubation with ACPA A (affinity purified), ACPA C (commercial), IgG or 16/6 Id. (C) Relative expression is shown following incubation of ACPA with Cit-ME or control peptide (Non-Cit-ME) for 2 h in room temperature prior to the addition of these antibodies to the cell culture. Results are presented as mean ± SEM of each group (CCP-positive RA, n = 9). *p ≤ 0.05; **p ≤ 0.0003; ***p ≤ 0.0001. ACPA: anticitrullinated protein antibodies; IgG: immunoglobulin G; CCP: cyclic citrullinated peptide; Cit-ME: multiepitope citrullinated peptide; Id: idiotype; NS: not significant; PBMC: peripheral blood mononuclear cells; RA: rheumatoid arthritis; RQ: relative quantification. SEM: standard error of the mean.
Synthetic peptides
Cit-ME with sequence V(Cit)L(Cit)SSVEST(Cit) GRS(Cit)PAPPPA(Cit)GLT, and its noncitrullinated control counterpart containing arginine instead of citrulline residues (Non-Cit-ME), were obtained from GL Biochem and described previously35. For ACPA blocking with Cit-ME, ACPA (400 ng/ml) was incubated with Cit-ME or with matched-control peptide Non-Cit-ME (100 ng/ml) for 2 h at room temperature before adding it to the cell culture. PBMC were cultured for 24 h in the presence of ACPA, ACPA with Cit-ME, ACPA with Non-Cit-ME, or IgG control alone. Thereafter, mRNA was isolated, and IL-1β and IL-6 gene expression were determined by real-time PCR.
Flow cytometry
For verifying surface binding of the experimental antibodies, 1 × 106 PBMC were suspended in 0.1 ml phosphate buffered saline. To block Fcγ receptors, cells were treated with Fc receptor–mediated antimurine CD16/32 cross react with human Fc receptors (eBioscience Inc.) for 20 min. Next, blocked PBMC were incubated with affinity purified ACPA (400 ng/ml), human IgG, or the 16/6 Id control at the same concentration for 1 h, after washing with PBS secondary antibody, FITC-labeled rabbit anti-human IgG was added for 1 h. The control cell sample was also incubated with the FITC-labeled rabbit anti-human IgG to measure the background florescence. Subsequently, 1 μl of propidium iodide 1 mg/ml was added to the samples. The acquired cells were analyzed in FITC channel for the mean fluorescence intensity (MFI) using FACScalibur flow cytometer (Becton Dickinson) and analyzed using FlowJo software (Tree Star).
Real-time PCR
RNA was isolated using Total RNA Purification Plus Kit (Norgen Biotek Corp.) according to the manufacturer’s instructions. RNA concentration was measured using Nanodrop (Thermo Scientific). For cDNA synthesis 1 µg total RNA was transcribed with cDNA transcription reagents using High Capacity cDNA Reverse Transcription Kit (Invitrogen) according to the manufacturer’s instructions. Gene expression was measured using real-time PCR performed on a StepOnePlus Real-Time PCR System (Applied Biosystems) according to the manufacturer’s instructions. Primer sequences (forward and reverse, respectively) were as listed in Table 1. The expression level of a gene in a given sample was represented as 2−ΔΔCt, where ΔΔCT = [ΔCT(experimental)] − [ΔCT(medium)] and ΔCT = [CT(experimental)] − [CT(housekeeping)]. The GAPDH levels were used to normalize gene expression levels.
Names, primer sequences, GenBank accession numbers, and amplicon lengths of specific genes used for real-time PCR.
Statistical analysis
Statistical analysis was completed using the 2-tailed, Mann-Whitney U test (GraphPad/Prism Version 5 software). Statistical significance was determined when p value < 0.05.
RESULTS
ACPA binding to peripheral blood cells from CCP–positive patients with RA and healthy controls
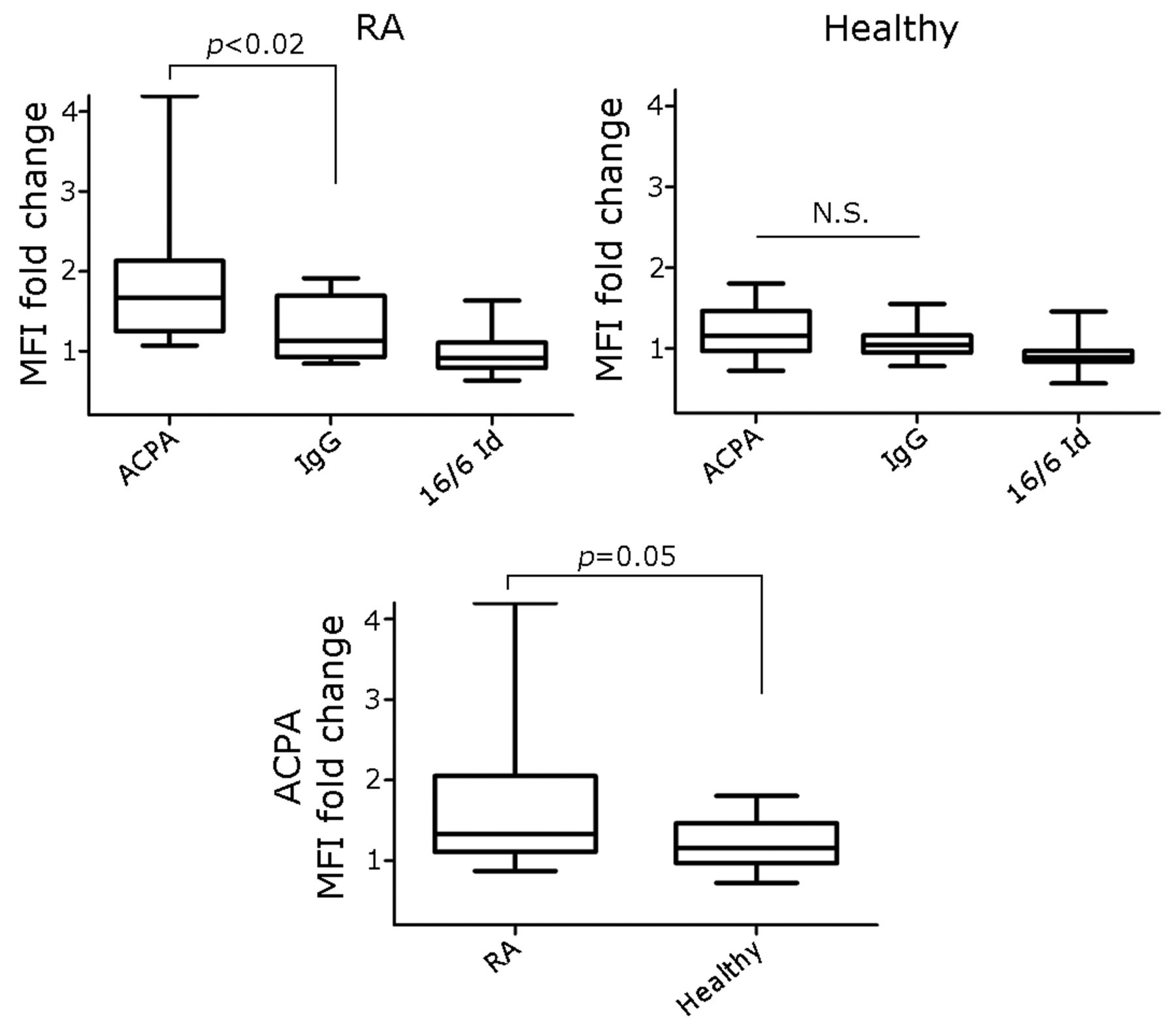
To determine whether ACPA bind PBMC of CCP–positive patients with RA and healthy controls, ACPA was incubated in vitro with PBMC from CCP–positive patients with RA (n = 18) and from healthy controls (n = 15). Cells were incubated with either ACPA or with the control antibodies; human IgG or an IgG carrying the 16/6 Id was analyzed by flow cytometry. Representative image of CCP–positive RA patient PBMC is shown in the left plot of Figure 2A; the right plot shows PI gating for exclusion of dead cells. Figure 2B shows histograms of PBMC stained with the experimental antibodies. Positive staining was determined by setting a marker defined with a negative unstained cell sample (not shown). While 1.1% of the cells were positive for secondary antibody alone, 3.5% of the cells were positive for ACPA, compared to 1.2% cells positive for irrelevant IgG and 0.8% positive for 16/6 Id. The overlay histogram image of ACPA versus irrelevant IgG is shown in Figure 2C. For each patient, MFI fold change was calculated by analysis of experimental antibody MFI divided by secondary antibody MFI. The analysis in Figure 3 shows that ACPA MFI fold change in CCP-positive RA patients’ PBMC were significantly higher (1.86 ± 0.22, p < 0.02) compared to irrelevant IgG and 16/6 Id (n = 18; 1.26 ± 0.09 and 0.97 ± 0.06, respectively). In healthy controls (n = 15), ACPA MFI fold change detected was 1.19 ± 0.08 (p = 0.26) compared to irrelevant IgG and 16/6 Id controls (1.07 ± 0.05 and 0.94 ± 0.05, respectively), but the differences were not significant. However, the MFI fold change of ACPA-positive PBMC from CCP-positive patients with RA (1.86 ± 0.22, p < 0.05) was significantly greater compared to healthy controls (1.19 ± 0.08).
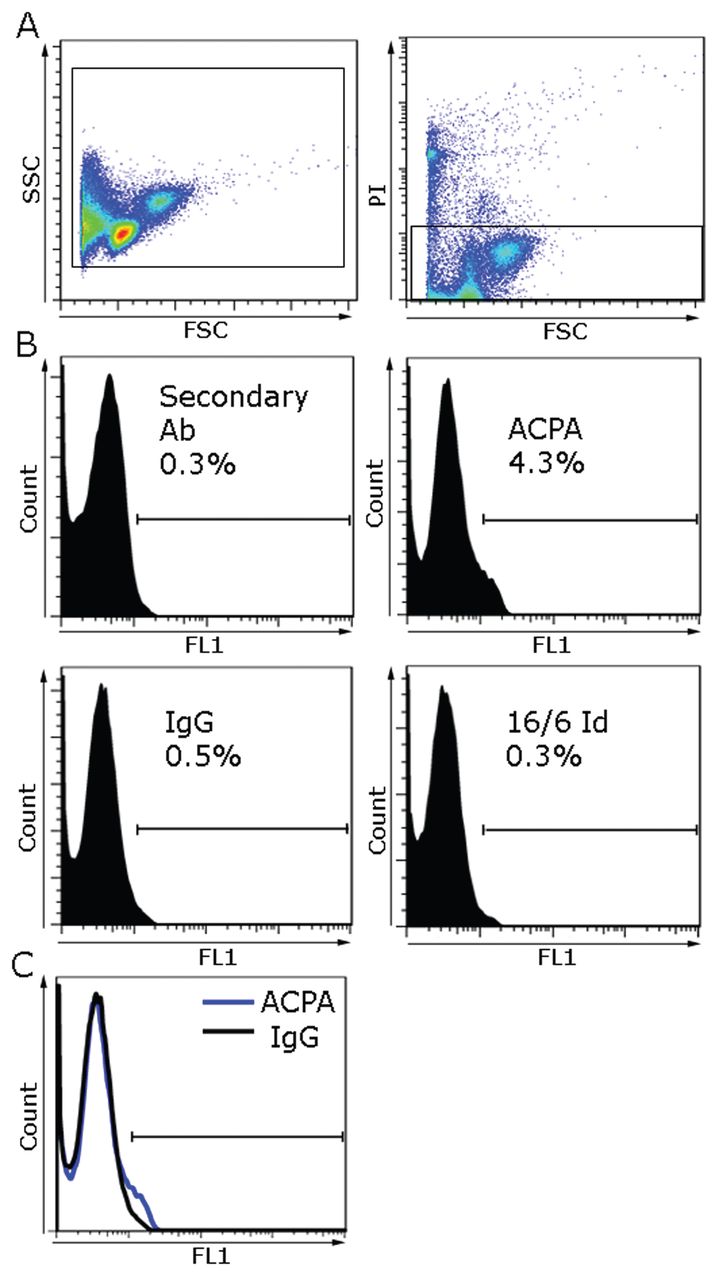
Representative analysis of PBMC derived from CCP-positive patient with RA positively stained with purified ACPA. (A) Left plot shows PBMC display on FSC versus SSC, and right plot shows gating for PI-negative cells. (B) Histogram of analysis for secondary Ab, ACPA, Human IgG, or Human 16/6 Id antibody-positive percentage of cells are shown by histogram marker in PBMC from representative CCP-positive patient with RA. (C) Overlay histogram represents ACPA detected on PBMC in FL1 Channel (blue line) or IgG (black line). Ab: antibody; ACPA: anticitrullinated protein antibodies; CCP: cyclic citrullinated peptide; FL1: fluorescence channel 1; FSC: forward scatter; IgG: immunoglobulin G; Id: idiotype; PBMC: peripheral blood mononuclear cells; PI: propidium iodide; RA: rheumatoid arthritis; SSC: side scatter.

MFI fold change of ACPA versus IgG and 16/6 Id detected on PBMC from CCP-positive patients with RA and healthy controls. Comparison of MFI fold change = (MFI of ACPA, IgG, or 16/6 Id)/(MFI of secondary Ab) in PBMC from CCP-positive patients with RA (n = 18) and healthy controls (n = 15) by flow cytometry analysis. Results are presented as mean ± SEM of each group, p values were obtained using the Mann-Whitney U test. MFI: mean fluorescence intensity; ACPA: anticitrullinated protein antibodies; IgG: immunoglobulin G; Id: idiotype; PBMC: peripheral blood mononuclear cells; CCP: cyclic citrullinated peptide; Ab: antibody; N.S.: not significant; RA: rheumatoid arthritis; SEM: standard error of the mean.
ACPA upregulate IL-1β and IL-6 expression in CCP–positive patients with RA
We further studied in vitro effects of ACPA on the inflammatory cytokines IL-1β and IL-6 mRNA expression in PBMC of CCP-positive patients with RA and healthy controls. PBMC were cultured for 24 h in the presence of ACPA (400 ng/ml) or the control antibodies at the same concentration. As shown in Figure 1, in vitro incubation of CCP–positive RA patients’ PBMC with ACPA significantly upregulated IL-1β and IL-6 gene expression (p < 0.0001) as compared with PBMC of the same patients cultured with the irrelevant IgG.
It should be noted that ACPA upregulated the IL-1β gene expression in PBMC of CCP–positive patients with RA up to 10-fold higher than that of the healthy controls (p < 0.0001). For IL-6, ACPA upregulated the expression in CCP-positive patients with RA to 4-fold that of the healthy controls (p = 0.0002; Figure 1A). In healthy controls, ACPA upregulated IL-1β expression (p = 0.003) and had no significant difference on IL-6 gene expression as compared to IgG [CCP-positive RA (n = 15) and healthy controls (n = 10)].
Figure 1B shows that in vitro incubation of the experimental affinity purified and commercial ACPA with PBMC from CCP–positive patients with RA resulted in upregulated IL-1β and IL-6 expression compared to irrelevant IgG, whereas the 16/6 Id control and the irrelevant IgG did not change the levels of these genes. Thus, these results suggest that ACPA are probably involved in the upregulation of IL-1β and IL-6 gene expression.
We further studied the possible inhibition of the ACPA effect on IL-6 and IL-1β expression by using Cit-ME. Its composition is based on a sequence juxtaposition to the citrullinated regions of the peptides that are considered highly reactive and commonly used in ACPA testing. We previously demonstrated the cross-reactivity of Cit-ME peptide binding to several ACPA (cit-filaggrin, cit-β-fibrinogen, cit-collagen, and cit-vimentin Ab) in an inhibition assay with ELISA35.
Figure 1C confirms the IL-1β and IL-6 upregulation by ACPA compared to the irrelevant IgG control in PBMC from CCP–positive patients with RA. To attenuate the ACPA-induced cytokines’ upregulation, Cit-ME or the control peptide, Non-Cit-ME, was added to ACPA before adding it to the culture. ACPA with Cit-ME inhibited upregulation of these genes by 30% for IL-1β (p = 0.05) and by 32% for IL-6 (p < 0.02). The control peptide, Non-Cit-ME, was less effective and its inhibition was not significant (IL-1β, p = 0.31 and IL-6, p = 0.22; CCP–positive RA, n = 9). Thus, inhibition of ACPA Fab could be involved in attenuating at least part of the effects induced by ACPA in arthritis.
ACPA alter expression of inflammatory mediator genes in CCP–positive patients with RA
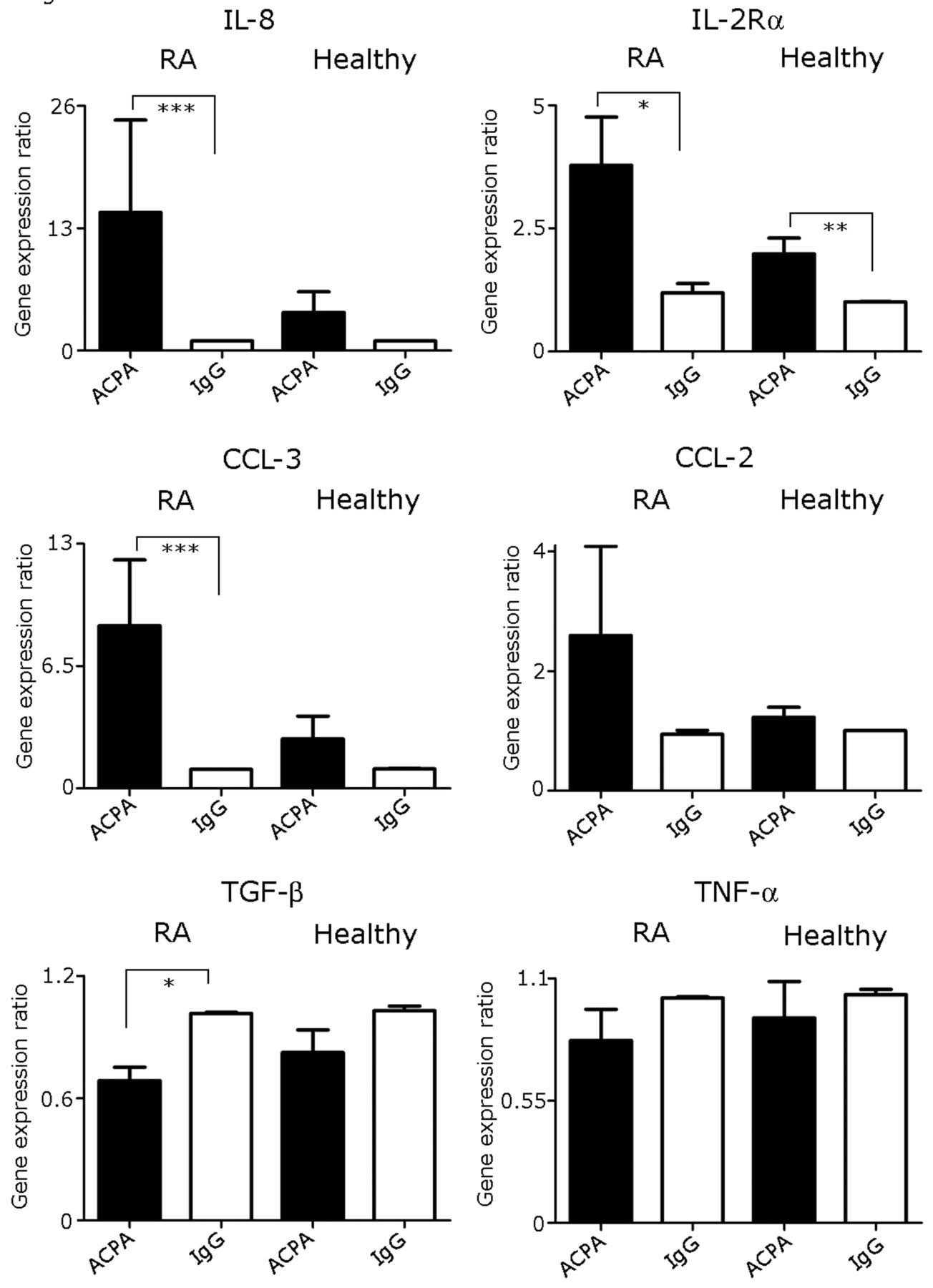
Since IL-1β and IL-6 were upregulated by ACPA, it was of interest to find out whether ACPA might alter the expression of other related genes. We measured cytokine gene expression for IL-8, IL-2Rα chain, CCL-3, CCL-2, TNF-α, and TGF-β. Figure 4 presents cytokine gene expression levels (normalization was performed to GAPDH and the irrelevant IgG control was considered as 1). In accordance with the expression pattern of IL-1β and IL-6, incubation of ACPA with PBMC of CCP–positive patients with RA resulted in significant upregulation of > 13-fold increase in IL-8 gene expression compared to incubation with IgG alone (p < 0.0001), while ACPA did not upregulate IL-8 expression compared to IgG control in PBMC of healthy controls.

Modulation of mRNA expression of cytokine mRNA expression profile in PBMC following coculture with ACPA. mRNA gene expression was detected by real time-PCR. Relative mRNA expression of each gene was normalized to GAPDH in PBMC from CCP-positive patients with RA (n = 10) and from healthy controls (n = 10). Results are presented as mean ± SEM of each group; *p < 0.002; **p = 0.004; ***p < 0.0006. ACPA: anticitrullinated protein antibodies; PBMC: peripheral blood mononuclear cells; CCP: cyclic citrullinated peptide; RA: rheumatoid arthritis; SEM: standard error of the mean; IgG: immunoglobulin G; TGF-β: transforming growth factor-β; TNF-α: tumor necrosis factor-α.
The IL-2Rα chain mRNA expression level was significantly elevated by ACPA, 4 times higher than the increment induced by the irrelevant IgG in CCP–positive patients with RA. In contrast, in healthy controls ACPA also significantly elevated IL-2Rα chain mRNA expression, but to a lesser extent (2× as much as the irrelevant IgG).
Likewise, ACPA significantly elevated the expression of CCL-3 mRNA in CCP–positive RA patients’ PBMC and not in healthy controls, as compared to IgG (p < 0.0006 and p = 0.48, respectively). CCL-2 mRNA expression levels were upregulated by ACPA as compared to the irrelevant IgG, but not significantly (p = 0.2). On the other hand, ACPA significantly downregulated the expression of the antiinflammatory cytokine TGF-β in CCP–positive RA patients’ PBMC compared to irrelevant IgG (p < 0.0015). ACPA did not significantly downregulate the level of the TGF-β gene expression in PBMC of healthy controls. However, ACPA did not significantly change TNF-α mRNA expression levels in PBMC of CCP–positive patients with RA or in healthy controls.
DISCUSSION
The main findings of our study are that affinity purified human ACPA could bind to a small fraction of PBMC derived from CCP–positive patients with RA. ACPA bind more PBMC of CCP–positive patients with RA than PBMC of healthy controls. We have shown that ACPA upregulated inflammatory cytokines mRNA molecules to a higher extent in CCP–positive RA patients’ PBMC than in normal controls.
ACPA binds a higher portion of PBMC derived from CCP–positive patients with RA following incubation with Fc blocker, compared to the control antibodies: irrelevant IgG or 16/6 Id as determined by MFI fold change (Figure 3). However, we cannot exclude the possibility that different treatments given to these patients with RA affected this binding.
Comparison of IL-1β and IL-6 mRNA levels in PBMC from CCP–positive RA patients with healthy controls revealed that ACPA upregulated both cytokines exclusively in PBMC from CCP–positive patients with RA. Elevated IL-1β and IL-6 mRNA expression were induced by both the affinity purified ACPA that targets 4 citrullinated autoantigens, and by commercial ACPA directed solely against citrullinated filaggrin (Figure 1B). Our study has certain limitations. Analysis of ACPA binding to PBMC was performed on PBMC blocked for Fc receptor. The latter was not blocked in the experiments testing change in cytokine genes expression, allowing ACPA to be bound through either the Fc or Fab domains. On the other hand, only ACPA (not IgG nor 16/6 Id) upregulated IL-1β and IL-6 expression in PBMC of CCP–positive patients with RA; in healthy controls, ACPA could not upregulate cytokines’ mRNA expression. One assumption is that ACPA binds through Fab that recognize citrullinated receptors on PBMC of CCP-positive patients with RA, similar to Grp78 expressed on human monocytes and macrophages. In those cells, upon ACPA binding, NF-κB and TNF-α are produced25,26. Moreover, this receptor is expressed on plasma cells of patients with RA and correlates ACPA levels36. However, our results still reveal a discrepancy since Cit-ME, which is the genuine ligand of the purified ACPA and neutralizes the Fab binding, could block only part of IL-1β and IL-6 mRNA upregulation (Figure 1C). Therefore, it remains to be identified which domain is responsible for ACPA-mediated effects by performing experiments of pepsin fragmentation to compare Fc and Fab activity separately.
Another hypothesis could be that ACPA possess specific characteristics, since only ACPA induce upregulation of IL-1β and IL-6 mRNA expression in CCP–positive RA patients’ PBMC, whereas IgG or 16/6 Id could not modify the expression levels of those inflammatory mediators (Figures 1 and 4). For illustration, the human monoclonal autoantibody 16/6 is a common anti-DNA idiotype found to have clinical relevance in patients with systemic lupus erythematosus (SLE)37,38. It was shown that the 16/6 Id induce proliferation and IL-2 secretion in SLE patients’ PBMC39. These results indicate that 16/6 Id moiety is pathogenic in the milieu of SLE, but not in that of RA. Each autoantibody could probably affect its own autoimmune environment, reflecting the specificities of their pathogenic roles. In this regard, it was shown that ACPA exhibit a specific Fc glycosylation profile that is distinct from other antibodies40 and Fc glycosylation phenotype could be involved in driving the disease process41.
ACPA increased IL-8 mRNA levels in only CCP–positive RA patients’ PBMC. IL-8 activates neutrophils which participates in inflammatory arthritis42. ACPA enhance osteoclast differentiation through IL-817. Also, mice injected with ACPA exhibit joint pain through osteoclast activation and release of IL-8 analog18. Here, ACPA upregulated IL-2α chain (CD25) in both CCP-positive patients with RA and in healthy controls’ PBMC. The IL-2 receptor is composed of 3 subunits (α, β, and γ); while the β and γ chains are constitutively expressed on T cells, the α chain is expressed on activated T cells43.
Elevated CCL3 (macrophage inflammatory protein-1α) levels and chemokine ligands CCL2 (monocyte chemoattractant protein-1) have been found in the serum and synovial fluid of patients with RA44,45. We showed that ACPA itself triggered the upregulation of CCL3 and of CCL2. Moreover, CCL3 and CCL2 are targets for citrullination by peptidylarginine deiminase enzyme in RA sera and synovial fluid46.
TGF-β was significantly reduced by ACPA in CCP–positive RA patients’ PBMC. Since TGF-β is an antiinflammatory cytokine, reduced TGF-β levels could further promote inflammation.
In our experiments, TNF-α levels were not significantly changed by ACPA. It was shown by Barbarroja, et al47 that ACPA could induce TNF-α mRNA expression in RA patients’ monocytes, but not lymphocytes, and Lu, et al showed that ACPA can promote TNF-α production in monocyte/macrophages25,26. Hence, the PBMC population in our cohort consisted most probably of lymphocytes, and ACPA is not likely to induce TNF-α expression in these cells. Alternatively, the time frame of TNF-α regulation in this assay could be different from that of the other genes tested.
ACPA are enriched in synovial fluid compared to serum48, and B cell clones specific for citrullinated proteins persist in the inflamed synovium of patients with RA49. In addition, a greater amount of peptidylarginine deiminase enzyme is released in the RA synovial fluid by neutrophils, which are responsible for the generation of new, local citrullinated autoantigens50. Based on these facts, we suggest that ACPA activity on synovial cells might be augmented, even compared to ACPA activity that was found here on peripheral cells. Yet, ACPA circulate in the blood (periphery) and its putative activity on peripheral cells was demonstrated here.
ACPA bind to PBMC of CCP–positive patients with RA and, to a lesser extent, to PBMC derived from healthy controls. In vitro, PBMC of patients with RA stimulated with ACPA upregulated inflammatory cytokines expression levels by 4– to 13-fold. A multiepitope citrullinated peptide inhibited part of ACPA cytokine upregulation, demonstrating that it is reasonable to suggest that ACPA neutralization could have therapeutic advantages.
Acknowledgment
The authors want to acknowledge Dr. Boris Gilburd for preforming the anti-CCP ELISA tests.
- Accepted for publication September 25, 2017.








{kind=link}
{kind=link}
{kind=link}
{kind=link}