

Abstract
Objective. Individuals with rheumatoid arthritis (RA) are at a heightened risk of sudden cardiac death, an outcome increased in those with prolongation of the corrected electrocardiographic QT interval (QTc). We compared QTc between patients with RA and demographically matched controls and studied the change in QTc after treatment with the interleukin 6 inhibitor tocilizumab (TCZ).
Methods. Standard 12-lead electrocardiograms were obtained and QTc was measured in patients with RA at baseline and after 24 weeks of TCZ treatment, then compared with non-RA controls who were frequency-matched on age and sex. Indicators of the baseline QTc and predictors of change in QTc were studied using multivariable linear regression.
Results. A total of 94 RA and 42 non-RA controls were studied. The average baseline QTc was 10 ms longer in the RA group vs controls (422 vs 412 ms, respectively; p < 0.001) and decreased to an average of 406 ms with treatment (p < 0.001). Baseline QTc was significantly and independently higher among those with anticyclic citrullinated peptide antibodies seropositivity, higher swollen joint counts, and higher levels of C-reactive protein (CRP) and matrix metalloproteinase 3. Each log unit decrease in CRP with treatment was associated with an average reduction in QTc of 2.9 ms (p = 0.002) after adjusting for age and baseline QTc. Clinical response measures were not associated with the change in QTc.
Conclusion. The marked normalization of QTc observed with TCZ treatment, and its close parallel with CRP reduction, support the premise that systemic inflammation contributes to cardiac repolarization abnormalities in RA that may be amenable to treatment.
Individuals with rheumatoid arthritis (RA) have a 2-fold higher risk of sudden cardiac death compared with age- and sex-matched controls1. Among the general (i.e., non-RA) population, several large studies have established that the risk of sudden cardiac death is increased in individuals with prolongation of the corrected QT (QTc) interval, as measured with electrocardiography (ECG)2,3. Chauhan, et al reported a significantly elevated risk of developing QTc prolongation among patients with RA4, and as reported by Panaoulas, et al, each 50 ms longer QTc was associated with a more than 2-fold higher hazard of all-cause mortality over an average followup time of 6 years5.
In many of these studies, circulating C-reactive protein (CRP)4,6 and/or circulating inflammatory cytokines7 were correlated with longer QTc intervals. These findings raise the possibility that RA treatments that reduce circulating levels of these offending cytokines may reduce the incidence of cardiac repolarization abnormalities that contribute to sudden cardiac death. However, whether QTc prolongation is reduced with treatment has not yet been well established. In a study of 17 patients with RA, Lazzerini, et al reported QTc normalization after treatment with the interleukin 6 (IL-6) inhibitor tocilizumab (TCZ), an effect that was correlated with the degree of reduction in CRP8. However, that study did not include comparison with a non-RA control group, and with the small sample size, only simple correlations of characteristics with QTc were possible. We sought to examine the QTc interval in a larger group of patients with RA compared with a demographically matched group of non-RA controls. Additionally, we sought to study the independent inflammatory and noninflammatory indicators of QTc in patients with RA and characterize the predictors of change in QTc associated with 24 weeks of treatment with TCZ. We hypothesized that clinical response to TCZ would be associated with a reduction in QTc in patients with RA, potentially returning to levels comparable to those of individuals without RA.
MATERIALS AND METHODS
Study population
Patients with RA were recruited from the Itabashi Chuo Medical Center from March 2012 to May 2016. Patients met the 2010 American College of Rheumatology/European League Against Rheumatism (EULAR) criteria for RA9, had moderate to severe RA disease activity based on the 28-joint count Disease Activity Score (DAS28) cutpoints10, and complied with the Guideline for Biological Products of the Japanese College of Rheumatology. Healthy volunteers were recruited as a control group to be frequency-matched to the age and sex distribution of the RA group. Both patients and controls were excluded if they reported prior cardiovascular (CV) events or procedures, whether self-reported or physician-diagnosed, including myocardial infarction, angioplasty, chronic heart failure, or arrhythmia. ECG exclusions included tachycardia (i.e., heart rate > 100 bpm), left bundle branch block, and ST elevations or depressions. The study was approved by the local ethics committee (Nihon University Itabashi Hospital RK-170912-12), and informed consent was obtained from each patient in accordance with the Helsinki Declaration of 1975 (revised in 1983).
Assessment and treatment protocol
Patients received TCZ either once a month (8 mg/kg intravenously) or 162 mg subcutaneously biweekly for 24 weeks. Electrocardiogram, clinical assessments, and laboratory monitoring were performed at baseline and Week 24. All patients were clinically evaluated by the same observer for disease activity measures. Low disease activity (LDA) was achieved if the DAS28 using CRP (DAS28-CRP) was < 3.2 units, and remission was achieved if the DAS28-CRP was < 2.6 units11. A EULAR good response was achieved if the DAS28-CRP achieved was < 3.2 units and the change from baseline was ≥ 1.2 units11.
ECG assessment
Resting 12-lead ECG (25 mm/s paper speed and 10 mm/mV amplitude) were recorded using a 3-channel direct writing machine at baseline and Week 24. The QT interval was corrected with the Bazett formula. A QTc ≥ 440 ms was considered prolonged, because this threshold has been demonstrated in several large studies to be associated with an increased risk of sudden cardiac death12,13. Accordingly, in this study the QTc interval of 440 ms was considered prolonged.
Clinical and laboratory assessments
Fasting samples of serum and plasma were collected at baseline and at Week 24, separated by centrifugation, and stored at −70°C. All assays were performed at our institution using our internal quality control procedures. At the time of the study visit, all subjects underwent routine laboratory investigations, and basic screening for traditional atherosclerotic disease risk factors, including history of cigarette smoking, serum cholesterol, triglycerides, high-density lipoprotein, low-density lipoprotein, fasting blood glucose concentration, rheumatoid factor, and anticyclic citrullinated peptide antibodies (anti-CCP). Collected samples were assessed blinded to group status or clinical characteristics. Matrix metalloproteinase (MMP-3) level was measured by the blood sample reacting with mouse antihuman MMP-3 monoclonal antibody in a turbidimetric immunoassay with sensitized latex-enhanced particles. CRP was measured by latex agglutination turbidimetric immunoassay.
Statistical analysis
Differences in participant characteristics according to disease status were compared using t tests for normally distributed continuous variables, the Kruskal-Wallis test for non-normally distributed continuous variables, and the chi-square goodness-of-fit test or Fisher’s exact test, as appropriate, for categorical variables. Linear regression was used to model predictors of baseline and change in QTc interval. Predictors were first modeled separately, and those with an association p value < 0.20 were carried into an extended multivariable (MV) model. Noncontributory covariates were excluded using Akaike Information Criterion for nested models. The adjusted R2 was used to assess the proportion of the variability in the outcome explained by the modeled covariates. The Shapiro-Wilk test was used to assess that normality assumptions for linear regression were met, which was the case for all the final MV models. Variance inflation factors were calculated to assess for collinearity among modeled covariates, and none was detected in all models. Statistical interaction of change in CRP with baseline QTc was tested in linear regression by introducing a QTc × change in CRP interaction term into the model. All statistical calculations were performed using Intercooled Stata 14 (StataCorp). A 2-tailed α of 0.05 was used throughout.
RESULTS
Baseline (pretreatment) characteristics of the 94 patients with RA and 42 non-RA controls are summarized in Table 1. The groups did not significantly differ on age or sex. Three-quarters of the patients had no CV disease (CVD) risk factors. Among those with CVD risk factors, the majority had hypertension (n = 14). Hyperlipidemia was present in 6 patients with RA, and only 1 had diabetes. RA disease activity was moderate to high, on average, and was reflected in above-normal markers of systemic inflammation in most. The majority of patients with RA (81%) were treated with disease-modifying antirheumatic drugs (DMARD), most prominently with methotrexate (MTX) at a median dose of 8 mg per week. Other DMARD (bucillamine, tacrolimus, and sulfasalazine) were uncommon (15%) and were used alone or in combination with MTX. Almost half were treated with prednisolone at baseline.
Baseline and followup characteristics. Data are median (interquartile range) unless otherwise indicated.
After 24 weeks of treatment, statistically significant improvements were observed in all disease activity measures, circulating inflammatory markers, and Health Assessment Questionnaire scores. Eighty-five percent achieved DAS LDA, 80% were in DAS remission, and 71% had no more than 1 swollen joint.
Elevated baseline QTc intervals decreased with TCZ treatment
The average baseline QTc was 10 ms longer in the RA group compared with controls (422 vs 412 ms, respectively; p < 0.001: Figure 1A). After treatment, the average QTc decreased to 406 ms in the RA group and was now significantly lower than the level of the control group. There were 12 RA patients with a baseline QTc ≥ 440 ms compared with none of the controls (p = 0.018, data not shown). After treatment, only 1 patient still had a QTc ≥ 440 ms. Treatment-associated reduction in QTc to levels at or below those of non-RA controls was observed in both men and women with RA (Figure 1B).
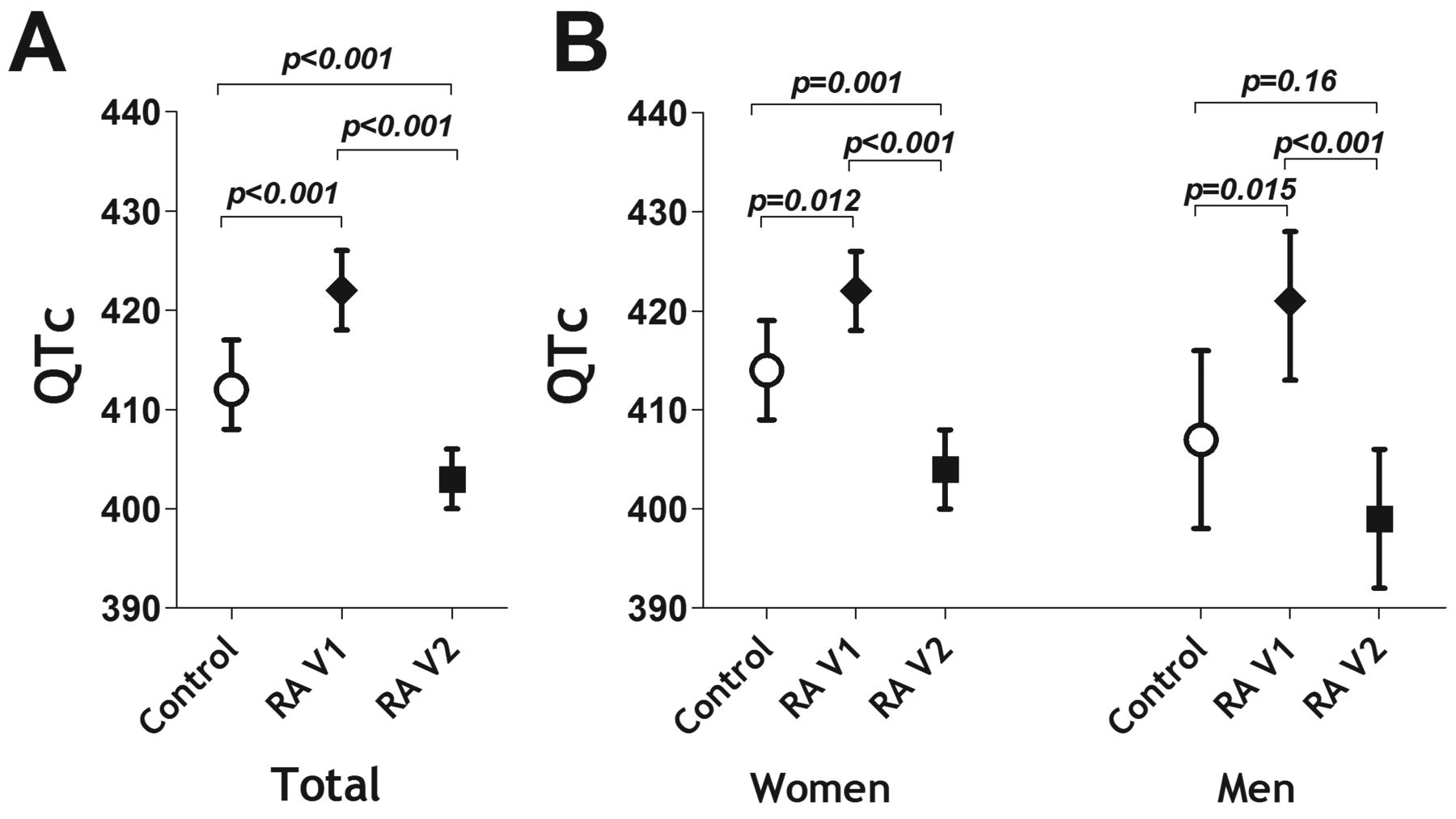
Comparison of QTc interval pre- and post-tocilizumab between RA patients and non-RA controls (baseline). Means and 95% CI are depicted. QTc: corrected electrocardiographic QT interval; RA: rheumatoid arthritis.
Baseline QTc interval was associated with anti-CCP seropositivity and multiple indicators of disease activity
Univariate and multivariable associations of RA patient characteristics with baseline QTc interval are summarized in Table 2. In the final model, those seropositive for anti-CCP had an adjusted QTc interval that was 8.3 ms longer than those who were seronegative (p = 0.021). On average, each additional swollen joint was associated with a 1-ms longer adjusted QTc (p = 0.038). Each log unit higher CRP was associated with a 3.7 ms longer adjusted QTc, on average, while each log unit higher MMP-3 was associated with a 5.2-ms longer adjusted QTc. Together, these 4 variables accounted for 26% of the explainable variability in baseline QTc interval (i.e., R2 = 0.255; p < 0.001). Neither demographics, the presence of CVD risk factors, nor baseline use of DMARD or prednisolone were significantly associated with baseline QTc.
Associations of patient characteristics with baseline QTc interval.
Patients above the median for swollen joint count (SJC; 4 joints), CRP (2.0 mg/dl), and MMP-3 (150 units) were classified as having the feature present. Those seropositive for anti-CCP and SJC, CRP, and MMP-3 all above the median had a mean baseline QTc of 436 ms compared with 412 ms for those without anti-CCP and SJC, CRP, and MMP-3 all below median levels (p = 0.003; Figure 2). Having any 1, 2, or 3 of these features was associated with progressively longer baseline QTc. Seven of the 22 RA patients with all 4 of the identified indicators of baseline QTc had a baseline QTc > 440 ms (32%) compared with none of those with none of the indicators (p = 0.029, data not shown).
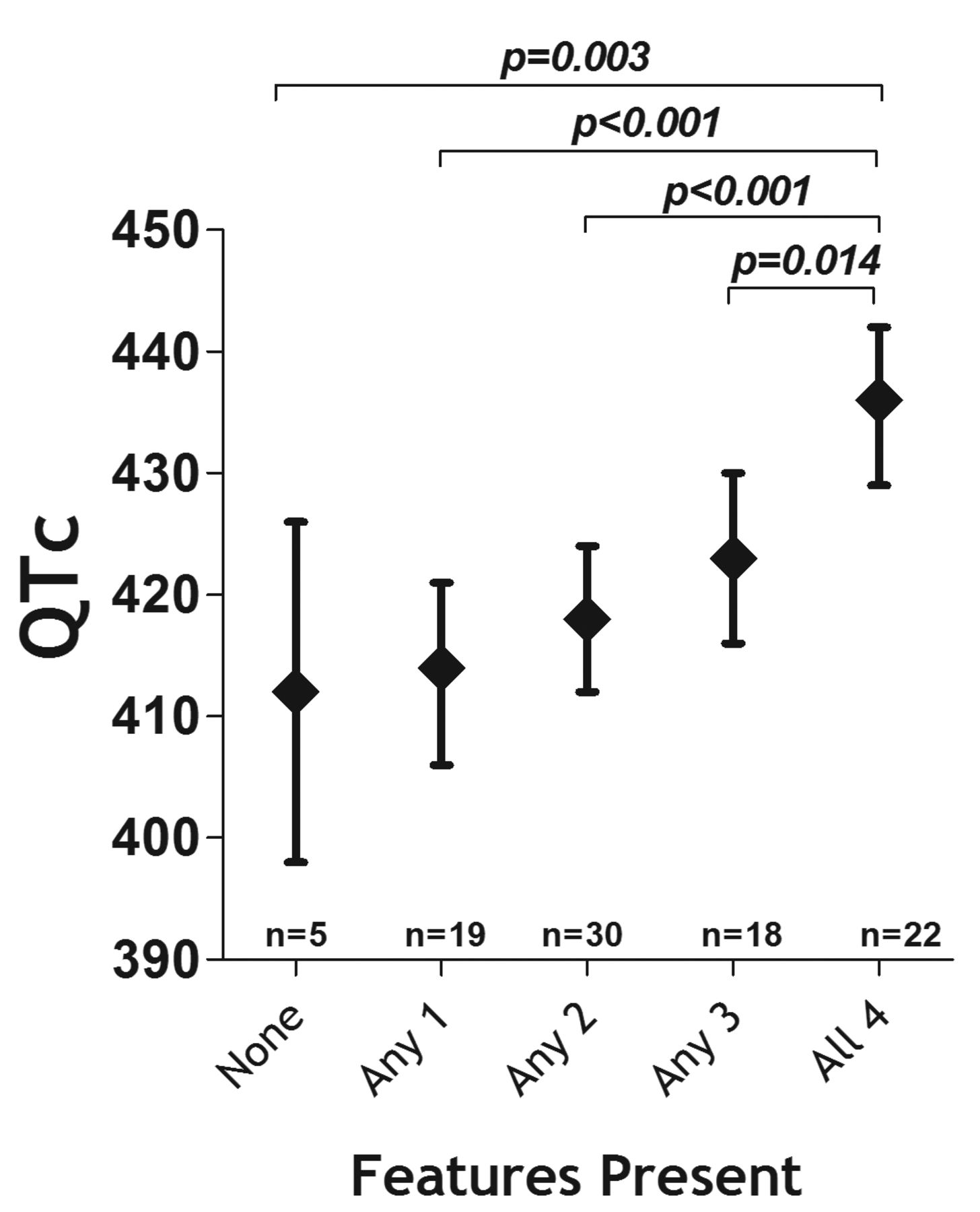
Adjusted baseline QTc according to the number of features present. Means and 95% CI are depicted. The features are the count of those identified from the reduced multivariable model depicted in Table 2. Presence for each feature was designated for anti-CCP > 20 units, swollen joint count above the median of 4 joints, C-reactive protein greater than the median of 2.0 mg/dl, and MMP-3 greater than the median of 150 units. Anti-CCP: anticyclic citrullinated peptide; MMP-3: matrix metalloproteinase 3; QTc: corrected electrocardiographic QT interval.
RA patients with the greatest reduction in CRP had the greatest reduction in QTc after TCZ treatment
Although a number of baseline characteristics were univariately associated with the change in QTc (Table 3, univariate models), only the change in CRP and the baseline QTc were significantly associated with the change in QTc when modeled together (Table 3, reduced multivariable model). Together, these 2 characteristics along with age accounted for 44% of the explainable variability in the change in QTc (i.e., R2 = 0.438; p < 0.001). Interestingly, clinical responders did not have a greater change in QTc compared with nonresponders independent of the change in CRP or baseline QTc, even when more stringent definitions of response were applied (i.e., DAS remission or having a maximum of 1 swollen joint).
Univariate and multivariable determinants of change in QTc.
Patients with RA who were in the highest tertile of CRP reduction (a reduction of ≥ 4.0 mg/dl) had a 26-ms decrease in QTc, on average, compared with a 14-ms reduction among those in the tertile with the smallest reduction in CRP (+0.09 to −1.0 mg/dl; Figure 3A), a difference that was significant (p = 0.003). Those in the highest tertile of baseline QTc had a nearly 3-fold larger reduction in QTc after treatment compared with those in the lowest tertile of baseline QTc (Figure 3B). Interestingly, among those with a baseline QTc ≥ 440 ms, each 1.0 mg/dl reduction in CRP was associated, on average, with a reduction in QTc of 11.2 ms (p = 0.002) compared with an average reduction in QTc of 3.4 ms for each mg/dl reduction in CRP among those with a baseline QTC < 440 ms (p = 0.001; p value for interaction = 0.039; Figure 3C).

Adjusted change in QTc interval after tocilizumab treatment according to change in CRP and baseline QTc interval. Means and 95% CI are depicted in panels A and B. Panel C depicts a more profound reduction in QTc according to the change in CRP among those with a higher baseline QTc interval (p value for interaction = 0.039). QTc: corrected electrocardiographic QT interval; CRP: C-reactive protein.
DISCUSSION
In this, to our knowledge, the largest study to date examining the effect of anticytokine therapy on QTc in RA, we observed a significantly higher QTc among a group of RA patients with a low prevalence of CV risk factors compared with a demographically matched group of non-RA controls. After 24 weeks of treatment with TCZ, QTc was reduced to levels that were significantly lower than those of the control group. The baseline QTc interval was strongly and independently associated with anti-CCP seropositivity, articular swelling, and several circulating inflammatory intermediates (CRP and MMP-3). However, only the change in CRP was a predictor of the change in QTc occurring with TCZ treatment. No other RA features were associated with the change in QTc independent of the change in CRP, including whether the patient had a clinical response to the drug. Interestingly, the association of the change in CRP with the change in QTc was markedly stronger among those with a prolonged baseline QTc.
Prolongation of the QTc interval is an established predictor of arrhythmia and sudden cardiac death in the general population, with moderate (QTc of 420–440 ms) and extensive QTc prolongation (> 440 ms) predictive of all-cause mortality13. Sudden cardiac death is also recognized as an important cause of death in patients with RA1; however, the pathophysiological mechanisms remain unclear. A strong link to systemic inflammation is suggested by several prior studies. Lazzerini, et al demonstrated a significant positive correlation between CRP and QTc duration in a cohort of 101 patients with chronic inflammatory arthritis6, and this association has been extended to circulating IL-6 levels14 and other inflammatory cytokines in RA7. How elevated circulating inflammatory cytokines prolong QTc is unclear, because they may affect the myocardium directly, or indirectly by increasing central nervous system sympathetic drive on the heart1.
Our study confirms these prior findings of a longer QTc associated with higher levels of circulating inflammatory markers, specifically CRP and MMP-3. We also found independent additive effects of SJC and seropositivity for anti-CCP on baseline QTc. Swollen joints are likely representative of additional unmeasured cytokines and inflammatory mediators, and indicative of additional inflammatory burden not determined by CRP or MMP-3. We also observed that anti-CCP–seropositive patients had longer QTc intervals. This could also represent a higher inflammatory burden in these patients, or there may be a direct link between antibodies against citrullinated proteins (ACPA) and the myocardium. In a prior publication15, we reported markedly higher staining for citrullinated proteins in the myocardial interstitium of patients with RA compared with non-RA controls and controls with systemic sclerosis and myocarditis. How ACPA may interact with the myocardium to prolong the QTc interval is unknown. However, the presence of anti-CCP did not impair the ability of TCZ to reduce QTc in our study after accounting for the change in CRP.
Our study also lends additional credence to the concept of IL-6 as a driver of QTc prolongation in RA, because targeted inhibition of IL-6 was associated with a marked reduction in QTc to levels that were even lower than those in the non-RA healthy population controls. This could be through direct inhibition of IL-6, or indirectly through the ability of IL-6 inhibition to reduce other cytokines and inflammatory mediators in RA. Our study expands on the findings of a pilot study8 in which a decrease in QTc was observed in 17 patients with RA after 12 weeks of TCZ treatment and was sustained to 24 weeks. Interestingly, this effect was independent of clinical response to TCZ, suggesting that the effect of TCZ on QTc is unrelated to its immunomodulating effect on synovitis. The ability of TCZ to reduce QTc to a level well below that found in healthy controls was somewhat unexpected. However, because the mechanism linking circulating IL-6 to QTc prolongation is likely not restricted to RA, it is possible, albeit speculative, that IL-6 inhibition in the general population may also have the effect of QTc reduction to the level we observed in the TCZ-treated patients with RA16.
The mechanism by which IL-6 inhibition reduced QTc is unclear. Previously, we reported that TCZ treatment significantly decreased left ventricular hypertrophy and normalized the aberrant left ventricular morphology that was associated with disease activity17. It is reasonable to posit that remodeling the myocardium to a more normalized structure may lead to QTc normalization. Additionally, an increasing body of evidence implicates macrophage-derived inflammatory cytokines, including tumor necrosis factor, IL-1, and IL-6, in electrophysiologic changes in cardiomyocytes, resulting in electrical remodeling of the heart that may predispose to prolonged QTc and ventricular arrhythmia (reviewed in Lazzerini, et al18). These include impaired potassium channel function19,20,21,22 and enhanced calcium channel function23,24 in cardiomyocytes, leading to prolongation of the cardiomyocyte action potential duration. Whether reduction of macrophage-derived cytokines with IL-6 inhibition is capable of reversing this electrical remodeling in RA warrants further study. Because the effect of TCZ on QTc was observed as early as 12 weeks in the pilot study by Lazzerini, et al8, it seems likely that the effect is mediated through electrical remodeling rather than structural remodeling, at least initially.
Our study has notable strengths and limitations. Among the strengths, the robust decline in QTc associated with decreasing CRP is a strong indicator of causality. However, without a comparison to untreated controls or a group treated with an immunomodulator targeting a different cytokine, we cannot assert that the observed normalization of QTc was not a natural phenomenon or that it is specific to IL-6 inhibition. In addition, IL-6 is the major cytokine determinant of hepatic CRP production. Thus, CRP may not be reflective of overall systemic inflammation in the setting of targeted IL-6 inhibition. While we focused on ECG as a marker of CVD, the ECG lacks specificity to detect early/preclinical myocardial lesions. We did not have additional imaging or advanced electrophysiological assessments to examine the mechanisms whereby IL-6 inhibition normalizes TCZ; however, based on our findings, such studies are certainly warranted and could prove useful not only to the study of arrhythmia in RA, but also for the general population. Finally, medications known to prolong QTc were not part of our data collection. However, for these to have impugned the differences detected they would have to have been markedly unbalanced between the RA and control groups and changed after TCZ treatment among enough patients with RA to normalize the QTc of the entire group. Such a scenario is unlikely to account for the magnitude of the differences and changes detected.
The marked normalization of QTc observed with TCZ treatment, and its close parallel with CRP reduction, support the premise that systemic inflammation contributes to cardiac repolarization abnormalities in RA that may be amenable to treatment, and suggest an antiarrhythmic effect of IL-6 inhibition that may be beneficial for patients with RA. Our data provide further evidence of the close link between RA autoimmunity, inflammation, and cardiac repolarization that may contribute to the known higher CVD risk in this population. Further investigation is warranted to evaluate the effect of TCZ on arrhythmia and sudden cardiac death in RA.
Acknowledgment
The authors thank all the clinical engineers, and the hematology and rheumatology staff at Nihon University School of Medicine Hospital for their assistance.
- Accepted for publication May 28, 2018.








{kind=link}
{kind=link}
{kind=link}