

Abstract
Objective. Age at onset has been shown to affect the clinical course and outcome of systemic lupus erythematosus (SLE). Herein, we aimed to define the differences in clinical characteristics, organ damage, and survival between patients with juvenile-onset (jSLE) and adult-onset SLE (aSLE).
Methods. For the study, 719 patients (76.9%) with aSLE and 216 (23.1%) with jSLE were examined. Comparisons between the groups were made for demographic characteristics, clinical features, auto-antibody profiles, damage, and survival rates.
Results. These results were significantly more frequent in jSLE: photosensitivity, malar rash, oral ulcers, renal involvement, neuropsychiatric (NP) manifestations, and autoimmune hemolytic anemia (AIHA). Of the autoantibodies, a higher frequency of anti-dsDNA and anticardiolipin IgG and IgM were observed in the jSLE group. A significant proportion of patients with aSLE had anti-Sm positivity and pleuritis. The proportion of patients with jSLE who developed organ damage was comparable to that of patients with aSLE (53% vs 47%) and the mean damage scores were similar in both groups. Renal damage was significantly more frequent in jSLE while musculoskeletal and cardiovascular system damage and diabetes mellitus were more prominent in aSLE. Comparison of survival rates of the 2 groups did not reveal any significant differences.
Conclusion. We report a higher frequency in the jSLE group of renal involvement, cutaneous symptoms, oral ulcers, NP manifestations, AIHA, and anti-dsDNA positivity. A significant proportion of patients in the jSLE group had damage, most prominently in the renal domain. Our findings might support different genetic/environmental backgrounds for these 2 subgroups.
Systemic lupus erythematosus (SLE) is a multisystemic chronic autoimmune disease that may cause a broad spectrum of clinical and immunological manifestations1. Along with ethnic and geographical influence, age at onset has also been shown to have a significant effect on the expression and outcome of the disease2,3,4,5. A direct correlation between the number of SLE susceptibility alleles and the risk of developing early-onset disease was described in a race-specific manner that was most pronounced in Gullah and African Americans6. In a Canadian cohort, 48% of the Asian patients were reported to have early-onset SLE with more aggressive disease, while 14% of white patients had juvenile-onset SLE (jSLE) and had a more benign disease with arthritis and mucocutaneous manifestations dominating7.
Patients with jSLE, who constitute nearly 20% of all patients with SLE, tend to have high disease activity at presentation, with an increased rate of organ involvement and longterm immunosuppressive treatment during the course of the disease8,9,10,11,12. Therefore patients with jSLE are more prone to develop disease and treatment-related damage, which may consequently lead to increased mortality9,10,13,14. Conceivably, these patients, when referred to adult rheumatology/SLE clinics, comprise a difficult group to manage with their high disease burden.
In most studies examining the differences between jSLE and adult-onset SLE (aSLE), a high prevalence of fever, lymphadenopathy, malar rash, and cytopenias has been found in jSLE15. Nephritis has also been reported to be more common in jSLE, without any significant differences in histologic subtypes10,13,14,16,17. Despite a lack of consensus on the frequency of occurrence of neuropsychiatric (NP) manifestations in jSLE, literature reviews and metaanalyses have shown that NPSLE is at least as common in patients with aSLE15,18. Reports on autoantibody profiles, serositis, and arthritis are conflicting, while mortality and damage data are scarce.
Herein, we aimed to compare clinical features, immunologic profile, damage, and survival rates of patients with jSLE and aSLE with different geographic backgrounds in a large combined longitudinal cohort of 2 tertiary centers in Istanbul.
MATERIALS AND METHODS
Patients
This analysis included 935 patients, 846 of whom attended the SLE outpatient clinic at Istanbul Faculty of Medicine between 1975 and 2011. The remaining 89 were followed in the pediatric rheumatology outpatient clinic at Cerrahpaşa Faculty of Medicine between 2004 and 2013. Overall, 719 patients had aSLE and 216 had jSLE. At the time of recruitment, all patients fulfilled the American College of Rheumatology (ACR) classification criteria for SLE19.
All patients registered were followed up by a standard protocol in both clinics. The protocol included data on demographic characteristics, SLE classification criteria, mortality, autoantibody profile, antiphospholipid syndrome (APS), features of nephritis including histopathology when available, and Systemic Lupus Erythematosus International Collaborating Clinics (SLICC)/ACR Damage Index (DI)19,20,21. APS was diagnosed according to the revised Sapporo classification criteria20. SLICC DI reflected the patients’ final visits. Duration of disease was defined as the time from the diagnosis of SLE to the time of last visit and duration of followup as the time from the first to the last visit of the patient in the SLE outpatient clinic. The data presented were the cumulative clinical and serological manifestations throughout the followup period. The study used the definition of the Pediatric Rheumatology International Trials Organization22 for jSLE: diagnosis at the age of 18 or younger. Demographic characteristics, clinical features, autoantibody profiles, and damage data were retrieved from the database and compared between the groups. All autoantibodies were tested at the immunology laboratory of our university in the routine clinical setting. Immunoblotting was used to detect anti-Sm, RNP, Ro, La, and anticardiolipin antibodies (aCL) IgG/M (EUROIMMUN Diagnostics), and anti-dsDNA was detected by immunofluorescence microscopy using Crithidia luciliae (INOVA Diagnostics). Positivity was confirmed at least twice for each patient at different timepoints. A positive test result for aCL was defined as IgM > 40 M phospholipid units and/or IgG > 40 G phospholipid units. Lupus anticoagulant (LA) was measured by the kaolin clotting time and/or dilute Russell’s viper venom time assays in the hematology laboratory.
The majority of patients with renal disease had histopathologically proven lupus nephritis (LN). Patients without renal biopsies either refused to be biopsied, had inconclusive biopsy results in which the tissue sample was insufficient for diagnosis, or had a contraindication to be biopsied. Biopsies were examined by experienced nephropathologists at Istanbul University, Istanbul Faculty of Medicine, Department of Pathology, and Cerrahpasa Faculty of Medicine, Department of Pathology and were classified by World Health Organization (WHO) before 2003 and International Society of Nephrology/Renal Pathology Society (ISN/RPS) 2003 systems afterward. Biopsies classified by WHO system were all reclassified by ISN/RPS system, and the results reflect this final classification.
As a followup procedure, all unattending patients were contacted by telephone. Patients who were nonresponsive to telephone calls and were not seen in the outpatient clinic within the last 6 months were considered lost to followup. The cause of death was extracted from patients’ hospital records or if unavailable, information was obtained from relatives contacted.
There was ethical approval to collect clinical information and blood from all our patients with SLE and APS. The patients gave written consent for the samples to be stored in our biobank and used in studies regarding SLE and APS [Istanbul University, Istanbul Faculty of Medicine, Clinical Research Ethical Committee (file: 2010/710-202, approval number: 852)].
Statistical analysis
Difference between the groups was analyzed by ANCOVA for continuous variables and by chi-square and logistic regression (LR) for categorical variables. The Kaplan-Meier method and log-rank test were used for survival analysis and Pearson’s correlation analysis to detect the strength of a link between 2 variables.
ANCOVA is a general linear model and an extension of ANOVA. It allows comparison of 1 variable in 2 or more groups correcting for the variability of 1 or more continuous variables, called covariates, that can affect the outcome23. To control for the duration of disease on the SLICC damage score, ANCOVA analysis was performed. LR is preferred to ANCOVA when the dependent variable of interest is binary. Thus, to explore independent associations between jSLE and aSLE groups and mortality, LR was used. All statistical analyses were performed using SPSS version 21. P values ≤ 0.05 were considered significant.
RESULTS
The total number of patients analyzed was 935. There were 216 patients (23.1%) with jSLE and 719 patients (76.9%) with aSLE. The proportion of female cases showed no significant difference when compared to male cases between the groups (87% vs 86% in jSLE and aSLE, respectively, p = 0.82). All patients studied were white. The mean age at SLE onset was 13.7 ± 3.5 years (range: 4–18 yrs) in jSLE and 34 ± 11.3 years (range: 19–72) in aSLE (p < 0.05). Both groups had a similar duration of followup with a mean of 79 ± 76 months (range: 6–366; median: 60 mos) in the jSLE group and 86 ± 76 months (range: 6–412; median: 67) in the aSLE group (p = 0.234). Duration of disease was significantly longer in patients with aSLE (111 ± 84 mos; range: 6–600; median: 102) compared to patients with jSLE (87 ± 96 mos; range: 6–396; median: 48; p = 0.001).
The prevalence of clinical features in both groups is outlined in Table 1. As in the previous cohorts, renal involvement was significantly more prevalent in the jSLE group, affecting 53.2% (n = 115) of the patients compared to patients with aSLE (38.9%, n = 280; p < 0.001). Autoimmune hemolytic anemia (AIHA; n = 72, 33.3% vs n = 68, 9.5%; p < 0.001) and NP involvement (n = 26, 12.1% vs n = 54, 7.6%; p = 0.04) also occurred more often in the jSLE group.
Cumulative clinical and laboratory findings. Data are n (%), unless otherwise indicated.
Table 2 shows the comparison of serologic profile in aSLE and jSLE groups. A higher proportion of patients with jSLE was positive for anti-dsDNA (n = 170, 78.7% vs n = 500, 69.5%, p < 0.009). Of the tested antiphospholipid antibodies, aCL IgG (n = 69, 31.9% vs n = 151, 21%) and IgM (n = 79, 36.6% vs n = 139, 19.3%) were more common in patients with jSLE despite the lack of a significant difference in the prevalence of APS (n = 26, 12% vs n = 111, 16%, p = 0.2 in jSLE and aSLE, respectively). Patients with aSLE had a higher frequency of anti-Ro (n = 184, 25% vs n = 23, 10.6%, p < 0.001) and anti-La antibodies (n = 81, 11.3% vs n = 11, 5.1%, p = 0.008) compared to patients with jSLE.
Prevalence of antibodies. Data are n (%), unless otherwise indicated.
Renal involvement affected 42% of the whole cohort, and 82% of these had biopsy-proven disease. Of 216 patients with jSLE, 115 (53.2%) had renal disease and 85 (74%) had a renal biopsy. Of the 719 patients with aSLE, 280 (38.2%) had LN, with 250 (89%) of them biopsy-confirmed. Histological analysis of LN subgroups demonstrated no significant differences, with diffuse proliferative LN dominating in both groups (Table 3).
Histopathologic subclasses of lupus nephritis (LN). Except for p values, data are n (%).
More than half of the patients with jSLE had damage according to the SLICC/ACR DI. The proportion of patients in the jSLE group with damage was higher than in the aSLE group, without any statistical significance in comparison (n = 112, 52% vs n = 326, 45% in jSLE and aSLE groups, respectively; p = 0.141) and the mean SLICC damage scores of patients with damage were similar in both groups (1.9 ± 1.2 vs 2.2 ± 1.6, p = 0.06, in jSLE and aSLE, respectively). No correlation was detected between the disease duration and SLICC damage score in the jSLE group, while a weak correlation was present in the aSLE group (r = 0.218, p < 0.05). Because Levene’s test for equality of variances was not significant (p > 0.05), a 1-way ANCOVA was conducted to more decisively determine whether a statistically significant difference existed between aSLE and jSLE mean SLICC scores in patients who had damage. ANCOVA results indicated that the duration of disease affected the SLICC damage score (p < 0.05). LR revealed that duration of disease significantly affected damage (p < 0.05). However, there was no significant effect of group type (jSLE or aSLE) on mean SLICC damage scores after controlling for disease duration (F = 1.094, p = 0.296).
When SLICC damage items were examined separately, renal damage was significantly more frequent in the jSLE (n = 49, 43.7%) compared to the aSLE group (n = 57, 17.4%; p < 0.001; Table 4). However, damage to the musculoskeletal system (avascular necrosis) was more prominent in the aSLE group (n = 101, 30.9% in the aSLE group vs n = 18, 16% in the jSLE group; p = 0.003). Cardiovascular system damage was also more prominent in the aSLE group (n = 77, 23.6% vs n = 14, 12.5%; p = 0.01). The number of patients with diabetes was significantly higher in the aSLE group (aSLE: n = 31, 9.5%; jSLE: n = 1, 0.9%; p = 0.003). Among 49 jSLE patients with renal damage, 12 (25%) had endstage renal disease (ESRD), 2 of whom received transplants. Four were receiving chronic hemodialysis, 16 (33%) had a more than 50% reduction in their glomerular filtration rate (GFR), and 36 (74%) had nephrotic range proteinuria exceeding 3.5 g/day. In the aSLE group, 48 (80%) of the 57 patients with renal damage had a GFR < 50%, 29 (48%) had nephrotic range proteinuria, and 26 (43%) had ESRD.
SLICC Damage Index. Data are n (%), unless otherwise indicated.
The main immunosuppressive drugs used were azathioprine, mycophenolate mofetil, cyclophosphamide, rituximab, and for a minority of patients, calcineurine inhibitors. Because this is a cohort including patients with different types of organ involvement, conceivably no single treatment protocol exists and the approach depends on the major organ(s) involved. Data on immunosuppressive usage were available in 80% of the cases. Owing to lengthy duration of disease and followup, data on immunosuppressive drugs in the early years for patients with jSLE were incomplete. However, the comparison after the transition to the adult outpatient clinic was insignificant and this may show that patients with jSLE still needed as much immunosuppression as patients with aSLE.
During the followup, there were 40 deaths, 8 of which were in the jSLE group and 32 were in the aSLE group. Table 5 lists the causes of death that could be identified in 29 patients. LR results showed that there was no relationship between mortality and aSLE and jSLE groups (p > 0.05) when disease duration was controlled. There were 112 patients (12%) lost to followup. Kaplan-Meier analysis showed survival rates of 98%, 92%, and 88% in the jSLE group and 97%, 93%, and 86% in the aSLE group at 5, 10, and 20 years, respectively (Figure 1). The mean duration of overall survival in jSLE and aSLE groups was 320 ± 14 months and 363 ± 9 months, respectively. Comparing the survival of 2 groups using the log-rank test did not reveal any significant differences (p = 0.8).
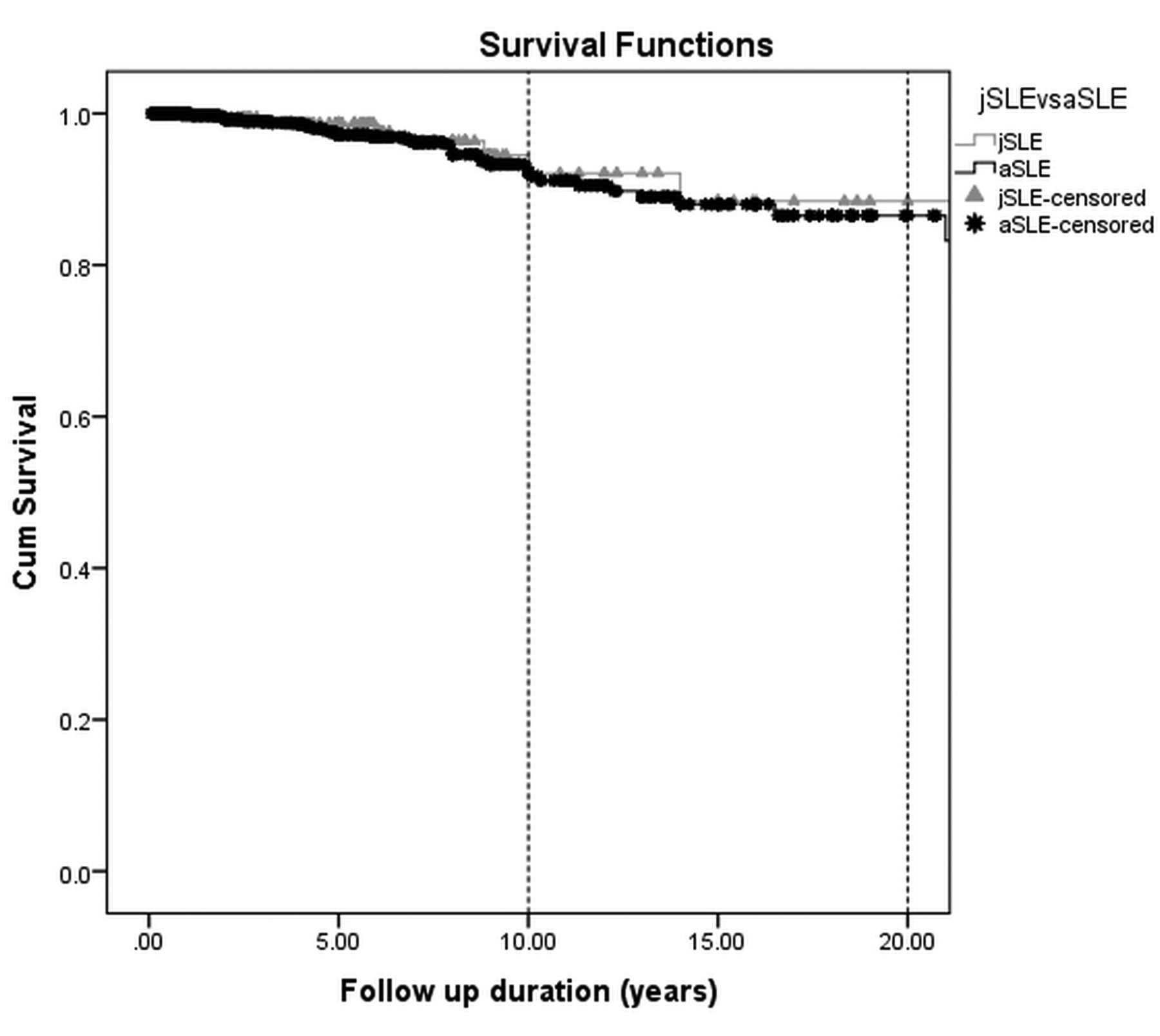
Kaplan-Meier survival curves of patients with jSLE and aSLE. The figure shows survival rates of 98%, 92%, and 88% in the jSLE group and 97%, 93%, and 86% in the aSLE group at 5, 10, and 20 years, respectively. Comparison of the 2 groups using the log-rank test did not reveal any significant differences (p = 0.8). jSLE: juvenile-onset systemic lupus erythematosus; aSLE: adult-onset SLE.
Causes of death in the whole cohort (total: 40; cause identified in 29).
DISCUSSION
Our study confirms that jSLE and aSLE are clinically and serologically different and that damage in patients with jSLE was as prevalent as in patients with aSLE. Examination of damage items separately displayed significant differences in some domains. Renal damage was more frequent in patients with jSLE.
SLICC/ACR DI is a validated instrument designed to measure irreversible damage in 12 organ systems resulting from SLE disease activity and its treatment and lasting for at least 6 months21,24. Increased organ damage due to active disease, lengthy duration of treatment, and comorbid processes is associated with increased mortality among patients with SLE, and early-onset disease may be a risk factor for greater damage and mortality25,26,27,28,29,30,31,32. In our cohort we found a high prevalence of damage in patients with jSLE despite their shorter duration of disease. Survival analysis showed similar rates in patients with jSLE and in patients with aSLE.
Among organ systems, renal domain was the most affected, both by active disease and damage in patients with jSLE. This finding is in line with previous studies17,18,33,34,35. Renal involvement is a major cause of mortality and morbidity in both aSLE and jSLE and carries a 10%–60% risk of ESRD36,37,38. Previously a more severe course has been shown in patients with early-onset SLE17,18,33,34,35. In a study in which late aSLE and jSLE patients were compared to non-SLE controls, patients with jSLE were found to have a greater risk of mortality and renal failure39. Among our 49 jSLE patients with renal damage, 12 had ESRD. Despite the similar distribution of renal histologic subtypes in both groups, the renal outcome supports a more aggressive course of disease in the jSLE group. Another explanation for this might be the insufficient intensity or late start of immunosuppressive treatment, leading to insufficient disease control. Although data on cumulative steroid doses are not available, a lower prevalence of damage that could also be attributed indirectly to steroid effect in patients with jSLE might indicate that high-dose steroid usage in this group is limited.
We found a higher prevalence of AIHA in patients with jSLE compared to patients with aSLE, as in other cohorts14,16,40. Previously, AIHA has been found to be associated with nephritis, NP involvement, thrombocytopenia, anti-dsDNA, and aCL antibodies in several cohorts14,38,39,40,41,42,43,44,45. These patients were described as having a more severe disease course, with higher disease activity and worse prognosis41,42,43,44. In our previous study, in which we defined autoantibody clusters and their clinical correlations and prognosis in our SLE cohort, we identified a cluster with aPL predominance (aCL IgG/IgM or LA), in which a significantly high rate of AIHA coexisted with thrombocytopenia, NP manifestations, and arterial and/or venous thrombosis46. Patients in this cluster had the highest frequency of damage and a significantly reduced survival at 10 and 20 years compared to other clusters. Damage was most pronounced in the NP domain because of arterial and venous vascular events. In our current study, despite the higher rate of occurrence of aCL IgG/IgM in patients with jSLE, the prevalence of APS was no different from that of patients with aSLE. Although positivity was confirmed at least twice in this cohort, it is important to keep in mind that young patients are more prone to viral infections, and nonpathogenic aPL can be detected. Further, the proportion of jSLE patients with LA positivity, which has the strongest correlation with vascular thrombosis, was less than that of patients with aSLE. The prevalence of NP involvement in the jSLE group was higher (despite the lack of significance) but left less damage compared to the aSLE group. One of the possible explanations could be the difference in the characteristics of NP events in the 2 groups of patients, with possibly temporary events dominating in the patients with jSLE.
Although our data originated from 2 dedicated centers, there were some limitations in our study. It had a retrospective design; our analysis was based on the cumulative findings, and progression of disease features by time could not be traced. Data on disease patterns were not available and information regarding treatment, which conceivably has a prognostic importance, was limited. Aside from the medications used and their cumulative doses, adherence to treatment should be an important issue to address in jSLE. Because data from 2 tertiary university hospitals were collected, a disproportionately higher number of severe cases may have been presented. All the patients included in this cohort are white and therefore their conclusions may not be applicable to other genetic groups.
However, this is the largest cohort study from Turkey of SLE patients with a longterm followup and with a limited number of patients lost to followup that also includes damage data, which have scarcely been reported.
The diseases jSLE and aSLE have different disease profiles. Patients with jSLE also have serious damage and have a marked mortality. Management differences and different genetic load in the juvenile- and adult-onset populations may be possible explanations for the differences observed. Considering the consequences of early-onset disease, a revision of the current treatment approach for this group of patients may be necessary. It is hoped that future molecular diagnostic techniques disclosing risk factors that predict a more severe course will make timely and appropriate treatment possible.
Footnotes
- Accepted for publication January 6, 2017.








{kind=link}