

Abstract
Objective. Apremilast, an oral phosphodiesterase 4 inhibitor, downregulates intracellular inflammatory mediator synthesis by elevating cyclic adenosine monophosphate levels. The PALACE 2 trial evaluated apremilast efficacy and safety in patients with active psoriatic arthritis (PsA) despite prior conventional disease-modifying antirheumatic drugs and/or biologic therapy.
Methods. Eligible patients were randomized (1:1:1) to placebo, apremilast 20 mg BID, or apremilast 30 mg BID. At Week 16, patients with swollen and tender joint count improvement < 20% entered early escape, with placebo patients rerandomized (1:1) to apremilast 20 mg BID or 30 mg BID while apremilast patients continued on their initial apremilast dose. At Week 24, patients remaining on placebo were rerandomized to apremilast 20 mg BID or 30 mg BID. The primary endpoint was the proportion of patients achieving > 20% improvement in American College of Rheumatology response criteria (ACR20) at Week 16.
Results. In the intent-to-treat population (N = 484), ACR20 at Week 16 was achieved by more patients receiving apremilast 20 mg BID [37.4% (p = 0.0002)] and 30 mg BID [32.1% (p = 0.0060)] versus placebo (18.9%). Clinically meaningful improvements in signs and symptoms of PsA, physical function, and psoriasis were observed with apremilast through Week 52. The most common adverse events were diarrhea, nausea, headache, and upper respiratory tract infection. Diarrhea and nausea generally occurred early and usually resolved spontaneously with continued treatment. Laboratory abnormalities were infrequent and transient.
Conclusion. Apremilast demonstrated clinical improvements in PsA for up to 52 weeks, including signs and symptoms, physical function, and psoriasis. No new safety signals were observed. ClinicalTrials.gov identifier: NCT01212757.
Psoriatic arthritis (PsA) is a chronic, systemic inflammatory disease that affects 0.3% to 1.0% of the general population1 and can lead to decreased physical function, impaired activities of daily living, and longterm disability2,3,4,5. Because of the disease’s chronicity, PsA treatment needs to be longterm, with the primary goals of maximizing health-related quality of life through control of symptoms and maintenance of patient function6. Current PsA therapy includes conventional synthetic disease-modifying antirheumatic drugs (csDMARD) and/or biologic agents7,8,9,10. Consistent efficacy in addressing the clinical manifestations of PsA has not been found with csDMARD8,10,11, which are associated with safety and tolerability concerns in some patients9,12,13. While many biologic DMARD (bDMARD) offer clinical and functional improvements for patients with PsA and affect radiographic progression of arthritis-related structural damage14,15,16, there are safety concerns that require monitoring17,18. Moreover, loss of efficacy and tolerability may limit longterm use of bDMARD for some patients19. New effective treatment options with an improved safety profile are needed for PsA.
Apremilast, an orally administered therapy, selectively targets phosphodiesterase 4 (PDE4) to downregulate the inflammatory cascade. PDE4 is an intracellular enzyme that helps regulate the expression of a network of proinflammatory and antiinflammatory mediators through degradation of cyclic adenosine monophosphate. In vitro, inhibition of PDE4 with apremilast has been shown to inhibit production of Th1, Th2, and Th17 cytokines20. In patients with PsA, apremilast treatment significantly decreased serum levels of proinflammatory cytokines interleukin 8 (IL-8), tumor necrosis factor (TNF)-α, and IL-6 during the first 24 weeks, and significantly decreased IL-6, IL-23, and IL-17 levels and increased antiinflammatory cytokines/biomarkers (IL-10, IL-1RA) over 40 weeks21.
The multinational Psoriatic Arthritis Longterm Assessment of Clinical Efficacy (PALACE) clinical development program, comprising 4 phase III randomized, placebo-controlled studies with open-label extensions, is evaluating apremilast in active PsA. PALACE 121, 2, and 322 assessed the efficacy and safety of apremilast 20 mg BID and 30 mg BID versus placebo in adult patients with active PsA despite prior treatment with csDMARD and/or biologics, including therapeutic failures, or concurrent csDMARD. These studies shared similar designs, except that PALACE 3 required ≥ 1 qualifying psoriasis lesion ≥ 2 cm. PALACE 2 included a higher proportion of European Union sites, which may have contributed to the lower proportion of patients in this study with prior bDMARD exposure. Our report describes results for PALACE 2 for up to 52 weeks of apremilast treatment.
MATERIALS AND METHODS
Study design
The phase III PALACE 2 study consisted of 3 treatment periods. In the placebo-controlled period, patients were randomized (1:1:1) to placebo, apremilast 20 mg BID, or apremilast 30 mg BID, stratified by baseline csDMARD use. The apremilast dose was titrated over the first treatment week, with increases of 10 mg/day until the target dose was reached.
Patients whose swollen joint count (SJC) and tender joint count (TJC) had not improved by ≥ 20% at Week 16 were defined as nonresponders and rerandomized (1:1) to apremilast 20 mg or 30 mg if initially randomized to placebo; if initially randomized to apremilast, treatment continued without a dose change. At Week 24, all patients who were still receiving placebo were rerandomized to apremilast 20 mg or 30 mg. Upon completing the 52-week blinded active-treatment period, patients could enter a longterm open-label phase of up to 5 years’ duration. Institutional review boards of the participating medical centers approved the study protocol, and all patients provided written informed consent before any study-related procedures were conducted.
Patients
Eligible patients were aged ≥ 18 years with a documented diagnosis of PsA with duration ≥ 6 months who met the Classification Criteria for Psoriatic Arthritis (CASPAR)23. Patients had to have ≥ 3 swollen and ≥ 3 tender joints despite prior treatment with csDMARD and/or bDMARD or concurrent treatment with csDMARD. TNF-inhibitor therapeutic failures were limited to ≤ 10% of randomized patients.
Patients with prior therapeutic failure of > 3 agents for PsA (csDMARD or bDMARD) or > 1 TNF inhibitor were ineligible. Patients could not have used phototherapy within 4 weeks, bDMARD (including adalimumab, etanercept, golimumab, infliximab, certolizumab pegol, or tocilizumab) within 12 weeks, or alefacept and ustekinumab within 24 weeks of randomization. Exclusions were prior apremilast treatment, active tuberculosis (TB), history of incompletely treated TB, or significant infection within 4 weeks of screening. No purified protein derivative or QuantiFERON screening for latent TB was required. There was no requirement to hold study medication for patients who developed an infection during the study and no prohibition on vaccinations in the protocol. Patients with erythrodermic, guttate, or generalized pustular psoriasis were excluded.
Concomitant medications
Patients were not required to be taking concurrent csDMARD therapy. Patients taking concurrent csDMARD at baseline could continue stable doses of methotrexate (MTX; ≤ 25 mg/week), leflunomide (≤ 20 mg/day), sulfasalazine (≤ 2 g/day), or a combination of these agents. A single reduction in a csDMARD dose was allowed between weeks 24 and 52. Patients could continue nonsteroidal antiinflammatory drugs if they were stable for ≥ 2 weeks before screening, and oral glucocorticoids (prednisone ≤ 10 mg or equivalent) if they were stable for ≥ 1 month before screening. These were permitted as background therapy, except ≤ 24 h before each study visit: low-potency topical glucocorticoids for treatment of face, axillae, and groin psoriatic lesions, coal tar shampoo and/or salicylic acid scalp preparations for scalp lesions, and nonmedicated emollient for body lesions.
Topical therapies for psoriasis, except those permitted for background therapy, were not allowed, including topical glucocorticoids, topical retinoids or vitamin D analog preparations, tacrolimus, pimecrolimus, or anthralin; immunosuppressive systemic therapy, including cyclosporine, oral retinoids, mycophenolate, thioguanine, hydroxyurea, sirolimus, azathioprine, and fumaric acid esters; and phototherapy (ultraviolet B, psoralen + ultraviolet A).
Efficacy assessments
The primary efficacy endpoint was the proportion of patients achieving an American College of Rheumatology (ACR20) response at Week 16, modified for PsA by including the first carpometacarpal joint and distal interphalangeal joint involvement of the feet to the total joint count24,25. Modified ACR20 response was defined as a ≥ 20% improvement from baseline in SJC and TJC, based on evaluation of 76 swollen and 78 tender joints, plus ≥ 20% improvement in ≥ 3 of these outcomes: (1) patient’s global assessment of disease activity [0–100 mm visual analog scale (VAS)], (2) physician’s global assessment (PGA) of disease activity (VAS), (3) patient’s assessment of pain (VAS), (4) Health Assessment Questionnaire–Disability Index (HAQ–DI), and (5) serum C-reactive protein (CRP) level.
The key secondary endpoint was change in HAQ-DI at Week 16. Additional secondary endpoints included proportions of patients achieving modified ACR50 and ACR70 responses, minimal clinically important differences (MCID) in HAQ-DI score (change from baseline ≥ 0.13 or ≥ 0.30) based on the literature at the time of protocol development26,27, 28-joint Disease Activity Score based on CRP (DAS28-CRP) < 2.6, Medical Outcomes Study Short Form-36 questionnaire (SF-36, version 2) physical functioning domain score, European League Against Rheumatism (EULAR) good or moderate response (good = DAS28-CRP ≤ 3.2 and improvement from baseline > 1.2; moderate = DAS28-CRP > 3.2 and improvement from baseline > 1.2, or DAS28-CRP ≤ 5.1 and improvement from baseline > 0.6 and ≤ 1.2), modified Psoriatic Arthritis Response Criteria (mPsARC), 50% and 75% reduction from baseline Psoriasis Area and Severity Index score (PASI-50 and PASI-75), and changes in individual ACR components and Clinical Disease Activity Index (CDAI) score. A posthoc analysis for the more recently published MCID ≥ 0.35 was also conducted28.
Safety assessments
Safety was evaluated at screening and weeks 0, 4, 16, 24, 28, 40, and 52 or at time of withdrawal based on adverse events (AE), vital signs, physical examination, and clinical laboratory measurements; a 12-lead electrocardiogram was obtained at screening and weeks 0, 16, 24, and 52. AE were classified using the Medical Dictionary for Drug Regulatory Activities classification system.
Statistical analysis
The sample size calculation was based on results from a phase II apremilast study29. A sample of 165 patients/group would be needed to yield 95% power to detect a 20% difference between apremilast treatment and placebo in ACR20 response, using a 2-group chi-squared test with a 2-sided 0.025 significance level.
Efficacy during the placebo-controlled period, including weeks 16 and 24, was evaluated for the intent-to-treat population, which included all patients who were randomized and received ≥ 1 dose of study medication. Sensitivity analysis was conducted for the per-protocol population, which included the intent-to-treat population with ≥ 1 post-baseline ACR evaluation and no critical protocol violations. Protocol violations excluding patients from the per-protocol population and all data-handling rules were determined before unblinding of the 24-week database. The primary endpoint, ACR20 response at Week 16, was analyzed using the Cochran-Mantel-Haenszel test, controlling for baseline DMARD use (yes/no); missing values were handled using the nonresponder imputation rule. Pairwise comparisons of each apremilast group versus the placebo group were performed using the Hochberg procedure to maintain type 1 error at 0.05. Results were considered statistically significant if both of the apremilast versus placebo comparisons achieved p < 0.05, or one of the comparisons achieved p < 0.025.
The key secondary endpoint, change from baseline HAQ-DI score at Week 16, was analyzed using an ANCOVA model, with treatment and baseline csDMARD use (yes/no) as factors and baseline value as a covariate; missing values were handled using the last observation carried forward (LOCF) methodology. Formal pairwise comparisons were conducted conditional on the test results of the primary endpoint and followed the Hochberg procedure, as described. Other binary and continuous variables were analyzed using the same methodology as described. Percent changes from baseline were analyzed based on the ANCOVA model using the rank transformation. For Week 24 analyses, patients who entered early escape at Week 16 were considered as having missing values at Week 24; the nonresponder imputation or LOCF imputed data were then applied as appropriate.
Efficacy analyses with no comparisons to placebo, including those at Week 52, were performed using observed data as prespecified.
Safety outcomes were analyzed among the safety population, comprising all randomized patients who received ≥ 1 dose of study medication. Results are presented for the placebo-controlled period (weeks 0–24), which included data through Week 16 for patients who initially received placebo and who escaped early, and data through Week 24 for all other patients, and for the 0- to 52-week period of apremilast exposure, which included all apremilast-exposure data, regardless of when treatment started (weeks 0, 16, or 24).
RESULTS
Patients
Overall, 484 patients were randomized and received ≥ 1 dose of study medication (Appendix 1). At Week 16, 55.3% (88/159) of patients receiving placebo, 36.2% (59/163) of patients receiving apremilast 20 mg, and 39.5% (64/162) of patients receiving apremilast 30 mg had < 20% improvement in SJC and TJC and entered early escape. A total of 428 (88.4%) patients completed Week 24, including those who escaped. Overall, 361 patients completed 52 weeks: 76.7% (125/163) of patients initially randomized to apremilast 20 mg, 70.4% (114/162) of patients initially randomized to apremilast 30 mg, and 76.7% (122/159) of patients randomized to placebo at baseline, including 60 patients receiving placebo/apremilast 20 mg and 62 patients receiving placebo/apremilast 30 mg (Appendix 1).
Patient demographics and disease characteristics were balanced across treatment groups (Table 1). The patient population was primarily white (95.0%), which is similar to populations in other PsA studies30,31. Similar proportions of patients in each treatment group were receiving csDMARD at baseline (69.8% to 71.1%), most commonly MTX. The mean (median; range) baseline MTX dose was similar in patients receiving placebo [14.6 mg/week (15.0; 5.0–25.0)], apremilast 20 mg [15.5 mg/week (15.0; 7.5–25.0)], and apremilast 30 mg [14.6 mg/week (15.0; 5.0–25.0)]. Similar proportions of patients in each treatment group had prior bDMARD exposure (14.2%–17.2%), and bDMARD had failed for 4.3% to 6.1%.
Baseline demographic and clinical characteristics: intent-to-treat population (N = 484). The “n” reflects the no. randomized patients who received at least 1 dose of study medication; actual no. patients available for each endpoint may vary slightly owing to missing data.
Primary efficacy endpoint: ACR20 response
At Week 16, a significantly greater proportion of patients receiving apremilast 20 mg (37.4%; p = 0.0002) and 30 mg (32.1%; p = 0.006) achieved an ACR20 response versus placebo (18.9%; Figure 1A). Sensitivity analysis, based on the per-protocol population, demonstrated consistent results (apremilast 20 mg: 38.4%, 61/159, p = 0.0002; apremilast 30 mg: 34.4%, 52/151, p = 0.0024; placebo: 19.5%, 30/154). In subgroup analyses, greater ACR20 response was seen at Week 16 with apremilast versus placebo, regardless of prior treatment experience. Higher absolute ACR20 response rates were observed in bDMARD-naive patients versus bDMARD-experienced patients and bDMARD failures. In bDMARD-naive patients, 39.3% of patients receiving apremilast 20 mg (53/135; p = 0.0009 vs placebo), 34.3% receiving apremilast 30 mg (46/134; p = 0.0130), and 20.7% receiving placebo (28/135) achieved an ACR20 response. In the small subpopulation of bDMARD-experienced patients, 28.6% receiving apremilast 20 mg BID (8/28), 21.7% receiving apremilast 30 mg (5/23), and 8.7% receiving placebo (2/23) achieved an ACR20 response. Greater ACR20 response was seen at Week 16 with apremilast versus placebo in patients with or without concomitant DMARD use. For patients receiving concurrent csDMARD treatment, ACR20 response was achieved by 41.2% (47/114) of patients receiving apremilast 20 mg (p = 0.0007), 36.6% (41/113) receiving apremilast 30 mg (p = 0.0079), and 20.4% (23/113) receiving placebo. A similar pattern, but lower response, was observed among patients not receiving concurrent csDMARD [apremilast 20 mg: 28.6% (14/49); apremilast 30 mg: 22.4% (11/49); placebo: 15.2% (7/46)]. The small sample size of the subgroup without concurrent csDMARD did not provide sufficient power to detect treatment differences.

(A) ACR20 in intent-to-treat population at Week 16 and (B) ACR20 by psoriatic arthritis subtype at Week 16. *p < 0.05, †p < 0.005 versus placebo. ‡Included predominant distal interphalangeal joint involvement, predominant spondylitis, and arthritis mutilans. ACR20: 20% improvement in modified American College of Rheumatology response criteria.
Subgroup analysis by PsA subtype showed similar ACR20 response rates at Week 16 in the apremilast-treated patients for asymmetrical oligoarthritis and symmetrical polyarthritis subtypes (Figure 1B). The number of individuals with predominant distal interphalangeal joints, arthritis mutilans, and predominant spondylitis was too small to analyze separately.
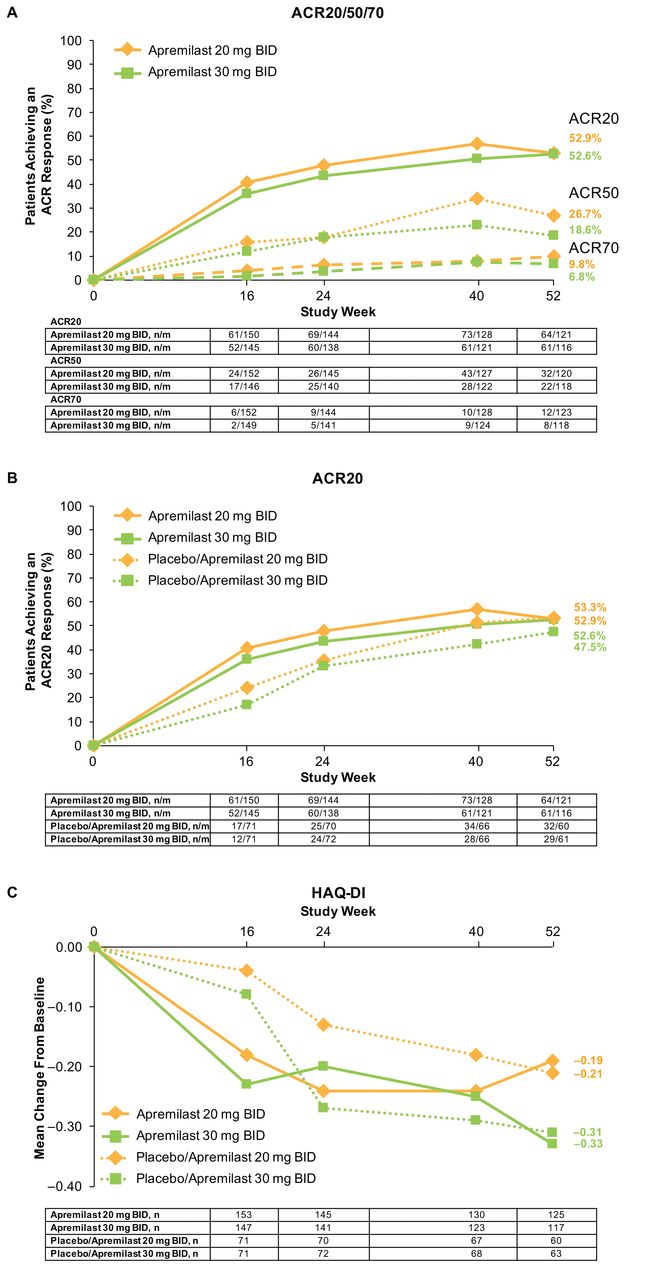
At Week 24, greater improvement in ACR20 response was seen with apremilast 20 mg and 30 mg versus placebo but not in ACR50 or ACR70 responses. In patients initially randomized to apremilast and remaining on treatment up to Week 52, overall ACR20 response was sustained over 52 weeks (Figure 2A, Figure 2B). At Week 52, 52.9% of patients initially randomized to apremilast 20 mg and 52.6% initially randomized to apremilast 30 mg achieved an ACR20 response (Table 2). Patients initially randomized to placebo who were rerandomized to apremilast demonstrated consistent results (Table 2; Figure 2B).

(A) ACR20/50/70, (B) ACR20, and (C) HAQ-DI over 52 weeks (data as observed). ACR20: 20% improvement in modified American College of Rheumatology response criteria; HAQ-DI: Health Assessment Questionnaire–Disability Index; n/m: no. responders/no. patients with sufficient data for evaluation.
Efficacy results at Week 52 (data as observed). The “n” reflects the no. patients who completed 52 weeks; actual no. patients available for each endpoint may vary. n/m = no. responders/no. patients with sufficient data for evaluation.
HAQ-DI
At Week 16, mean change from baseline in HAQ-DI score was significantly greater with apremilast 20 mg (−0.17, p = 0.032) and 30 mg (−0.23, p = 0.0042) versus placebo (−0.07; Table 3). The per-protocol population sensitivity analysis was consistent with the primary analysis (apremilast 20 mg: −0.17, p = 0.0319; apremilast 30 mg: −0.23, p = 0.0047; placebo: −0.07). At Week 24, mean change in HAQ-DI from baseline was significantly greater with apremilast 30 mg (−0.24, p = 0.0191) versus placebo (−0.10). Improvement with apremilast 20 mg (−0.17) was not significantly different from placebo (−0.10).
Secondary outcomes at Week 16 (intent-to-treat population). The “n” reflects the no. randomized patients who received ≥ 1 dose of study medication; actual no. patients available for each continuous endpoint may vary slightly owing to patients without a post-baseline value at or before Week 16.
Improvements in HAQ-DI scores were observed in patients initially randomized to apremilast and completing 52 weeks (Figure 2C). At Week 52, mean changes in HAQ-DI scores were −0.19 (apremilast 20 mg) and −0.33 (apremilast 30 mg; Table 2). A total of 36.8% (46/125) of patients initially randomized to apremilast 20 mg and 47.0% (55/117) initially randomized to apremilast 30 mg achieved an MCID ≥ 0.35.
Additional efficacy endpoints
Efficacy of apremilast versus placebo at Week 16 was seen across a number of endpoints (Table 3). Therapeutic effect continued through Week 24, including improvements in ACR20 response, HAQ-DI (apremilast 30 mg), SF-36v2 physical functioning domain (apremilast 30 mg), good/moderate EULAR response, mPsARC response (apremilast 20 mg), DAS28-CRP, CDAI, SJC, TJC, CRP (apremilast 30 mg), PGA, PASI-50, and PASI-75. At Week 52, improvements in efficacy measures were observed among patients initially randomized to apremilast and completing 52 weeks, and among those initially randomized to placebo and then randomized to apremilast at Week 16 or 24 (Table 2), including ACR20 response, HAQ-DI, SJC, TJC, PASI-50, and PASI-75.
Safety and tolerability
During the placebo-controlled period (weeks 0–24), the most common AE (occurring in ≥ 5% of any treatment group) were diarrhea, nausea, headache, and upper respiratory tract infection. The nature, incidence, and severity of AE were comparable over 24 and 52 weeks (Table 4). AE leading to discontinuation were < 10% for patients exposed to either apremilast dose up to Week 52. During the 52-week apremilast-exposure period, 23 patients (5%) experienced a serious AE [(SAE); apremilast 20 mg: n = 11 (5%); apremilast 30 mg: n = 12 (5%)]. All individual SAE were reported for 1 patient each, except depression, hypertension, and psoriatic arthropathy, which were reported for 1 patient in each treatment group. One patient receiving apremilast 20 mg developed an SAE of small intestine diverticulitis leading to treatment interruption, which resolved with intravenous antibiotics. The study drug was resumed after hospitalization without further episodes of intestinal diverticulitis.
Adverse events: placebo-controlled (Weeks 0 to 24) and apremilast-exposure (Weeks 0 to 52) periods.
Abnormalities in clinical chemistry and hematology variables were infrequent, with isolated changes meeting criteria to be markedly abnormal (Table 4). Most abnormalities were single values outside of normal ranges with no trends observed; patients continued apremilast treatment with no further changes in laboratory variables.
Diarrhea and nausea
During the placebo-controlled period, diarrhea and nausea were predominantly mild to moderate in severity. Mild/moderate and severe diarrhea incidence rates reported during this period were 5.0% and 0.0%, respectively, with placebo, and 12.6% and 0.3%, respectively, with apremilast. Mild/moderate and severe nausea incidence rates were 1.9% and 0.0%, respectively, with placebo, and 12.0% and 0.6%, respectively, with apremilast. A small number of patients taking apremilast reported using drug treatments directed toward these symptoms. Diarrhea and nausea generally occurred during the first 2 weeks of treatment, although there were occurrences throughout the study, and they were usually resolved within 1 month despite continued treatment and without medicinal intervention. Discontinuations due to diarrhea and nausea in the combined apremilast treatment group were < 2% each over 52 weeks.
AE of interest
No serious opportunistic infections, systemic vasculitis, major adverse cardiac events, or deaths occurred during the study. Patient-reported TB-related medical history included latent TB (n = 3), pulmonary TB (n = 1), and TB (n = 1). No reactivation of TB or cases of de novo TB were reported up to Week 52. One patient (a 30-year-old man), who was initially randomized to placebo and then rerandomized to apremilast 20 mg, was diagnosed with T cell lymphoma after 29 days of apremilast treatment; he was withdrawn from the study. Tissue diagnosis of lymphoma was not confirmed after repeated biopsies. During the apremilast-exposure period, a 58-year-old woman randomized to apremilast 20 mg developed basal cell carcinoma, which was reported as an SAE and led to treatment interruption for only several days.
Body weight
Weight decrease was reported as an AE in several patients during the 52-week apremilast-exposure period [apremilast 20 mg: 1 patient (0.4%); apremilast 30 mg: 3 patients (1.3%)]. Most patients maintained their weight within 5% of baseline; weight loss > 5% was observed in 39 patients (17.0%) exposed to apremilast 20 mg and 34 (14.8%) exposed to apremilast 30 mg. No patients with > 5% weight loss experienced clinical sequelae that can occur with weight loss, including dehydration, electrolyte disturbances, malnutrition, or cholelithiasis. No patients experienced weight loss > 20%.
DISCUSSION
In this phase III, placebo-controlled study, oral apremilast demonstrated clinical improvements in signs and symptoms, psoriasis, and physical function in patients with active PsA for up to 52 weeks. At the primary endpoint, modified ACR20 response at Week 16, both apremilast doses demonstrated statistically greater efficacy versus placebo, regardless of prior treatment, although only about 15% of patients with prior bDMARD exposure were randomized.
Escape criteria had low thresholds, mandating that patients enter rescue treatment early. An analysis of patients randomized to apremilast at study onset who qualified for early escape at Week 16 showed that about 30% in each dose group achieved a modified ACR20 response at Week 24, suggesting that a subset of patients may require more time to achieve response (data not shown). Trends for improvements across multiple facets of PsA were observed through Week 52.
At Week 16, apremilast 20 mg was associated with a higher modified ACR20 response versus apremilast 30 mg. However, by Week 52, response rates were similar for both dose groups. Looking at the PALACE program as a whole, higher responses were generally observed for apremilast 30 mg than 20 mg at Weeks 16 and 24, with comparable AE rates for both doses.
Both apremilast doses were generally well tolerated. The safety profile was similar to that reported previously with apremilast22,28,32,33,34, and comparable for the 24-week and 52-week periods. Diarrhea and nausea, the most common AE, were predominantly mild, usually occurred early, and generally resolved with continued use, and notably, without medicinal intervention. No clinically meaningful trends were observed in vital signs or laboratory abnormalities; findings did not indicate a need for laboratory monitoring.
Efficacy over longer-term studies can be biased, because patients not responding to or tolerating therapy are more likely to discontinue. In this 52-week study, 7.1% of patients receiving apremilast at baseline discontinued because of lack of efficacy, and 7.4% discontinued because of AE. PALACE 2 patients were required to have prior treatment with csDMARD and/or bDMARD, and these results may not be comparable to patients who are treatment-naive. Although this study included individuals with prior bDMARD exposure, that subpopulation was small; thus, results reported here may not be generalizable to that patient population.
The PALACE 2 findings support available data demonstrating that apremilast is effective for the treatment of active PsA by improving signs and symptoms of PsA, physical function, and severity of psoriasis with up to 52 weeks of treatment. This study did not reveal any new safety signals, showing that apremilast is safe without the need for laboratory monitoring. The benefit:risk profile and oral route of administration suggest that apremilast may represent an effective, alternative treatment option for patients with active PsA.
Acknowledgment
The authors received editorial support in the preparation of this report from Jennifer Schwinn, RPh, and Kristin Carlin, RPh, MBA, of Peloton Advantage LLC and funded by Celgene Corp.
APPENDIX 1.
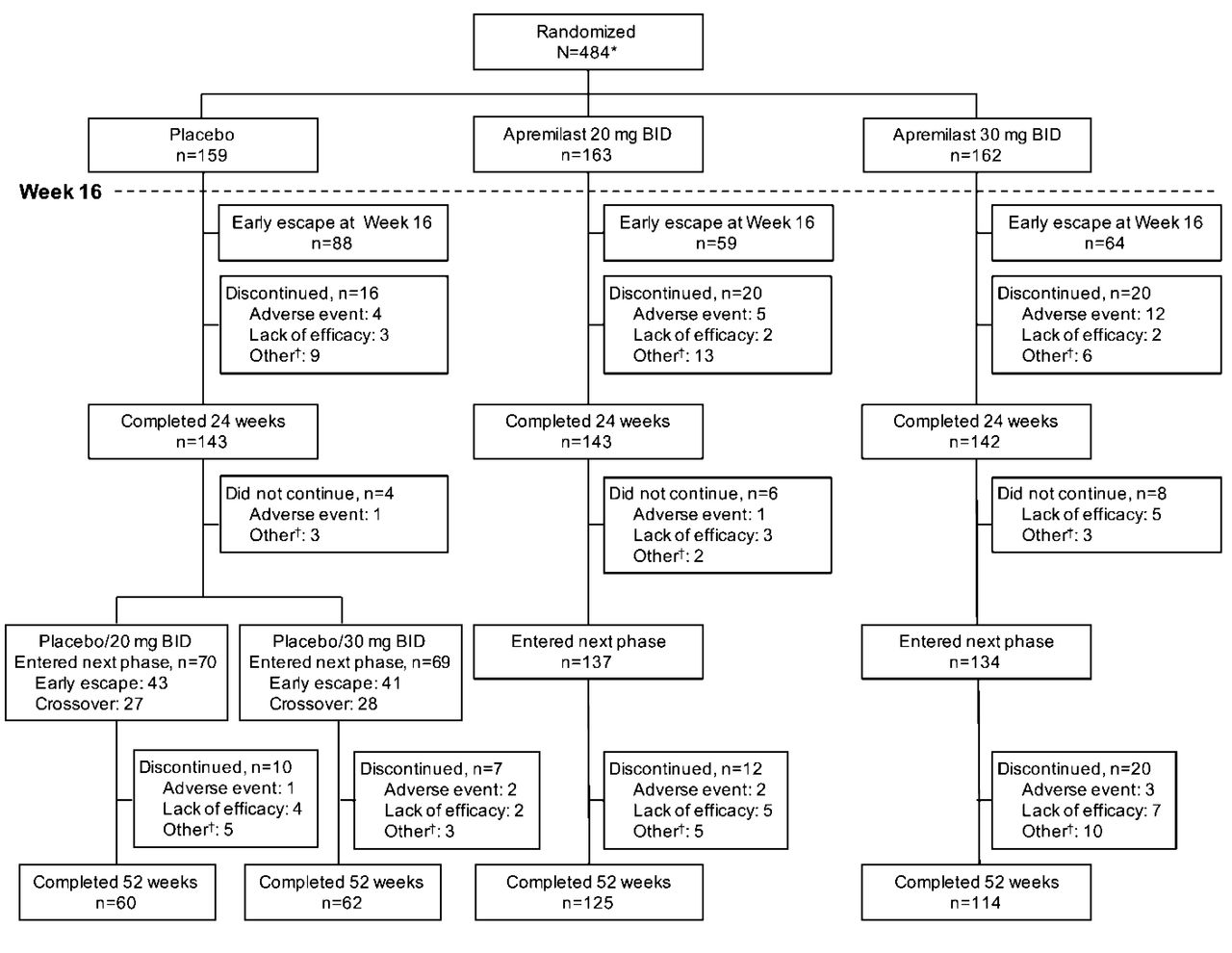
Patient disposition through Week 52. *Excludes 4 patients who were randomized in error and did not receive any dose of study medication. †Includes withdrawal by patient, loss to followup, protocol violation, noncompliance, and other.
Footnotes
Study sponsored by Celgene Corp. Editorial support for this article funded by Celgene. The authors are fully responsible for all content and editorial decisions. M. Cutolo has received research grants and/or consulting fees from Actelion, Bristol-Myers Squibb, Mundipharm, and Sanofi-Aventis. G.E. Myerson has received research grants and/or consulting fees from AbbVie, Actelion, Amgen, Bioventus, Bristol-Myers Squibb, GlaxoSmithKline, Eli Lilly, Pfizer, Primus, Roche (Genentech), Takeda, and UCB. These authors have received research grants and/or consulting fees from Celgene Corp.: R.M. Fleischmann, F. Lioté, F. Díaz-González, and F. Van den Bosch. E. Feist has received research grants and/or consulting fees from Bristol-Myers Squibb, Eli Lilly, and Novartis. K. Shah and C. Hu are employees of Celgene Corp. R.M. Stevens is a former employee of Celgene Corp.
- Accepted for publication May 31, 2016.








{kind=link}
{kind=link}
{kind=link}