

To the Editor:
Neuromyelitis optica spectrum disorders (NMOSD) are a group of rare autoimmune diseases typically affecting women in their 40s and 50s, as does Sjögren syndrome (SS), another autoimmune disease. We describe a 6-year-old girl who presented with clinical and laboratory findings of both an NMOSD and SS.
A 6-year-old previously healthy Korean girl presented with a 1-day history of fever, headache, progressive right-sided weakness, and altered mental status. Cerebrospinal fluid (CSF) analysis revealed an elevated protein without pleocytosis or oligoclonal bands. Magnetic resonance imaging (MRI) of the spinal cord showed contiguous T2 hyperintensity, and MRI of the brain showed extensive white matter lesions bilaterally (Figure 1), suspicious for demyelinating disease. She was treated with high-dose methylprednisolone, resulting in marked improvement, and continued treatment with tapering doses of prednisone, but flared when the prednisone was discontinued.
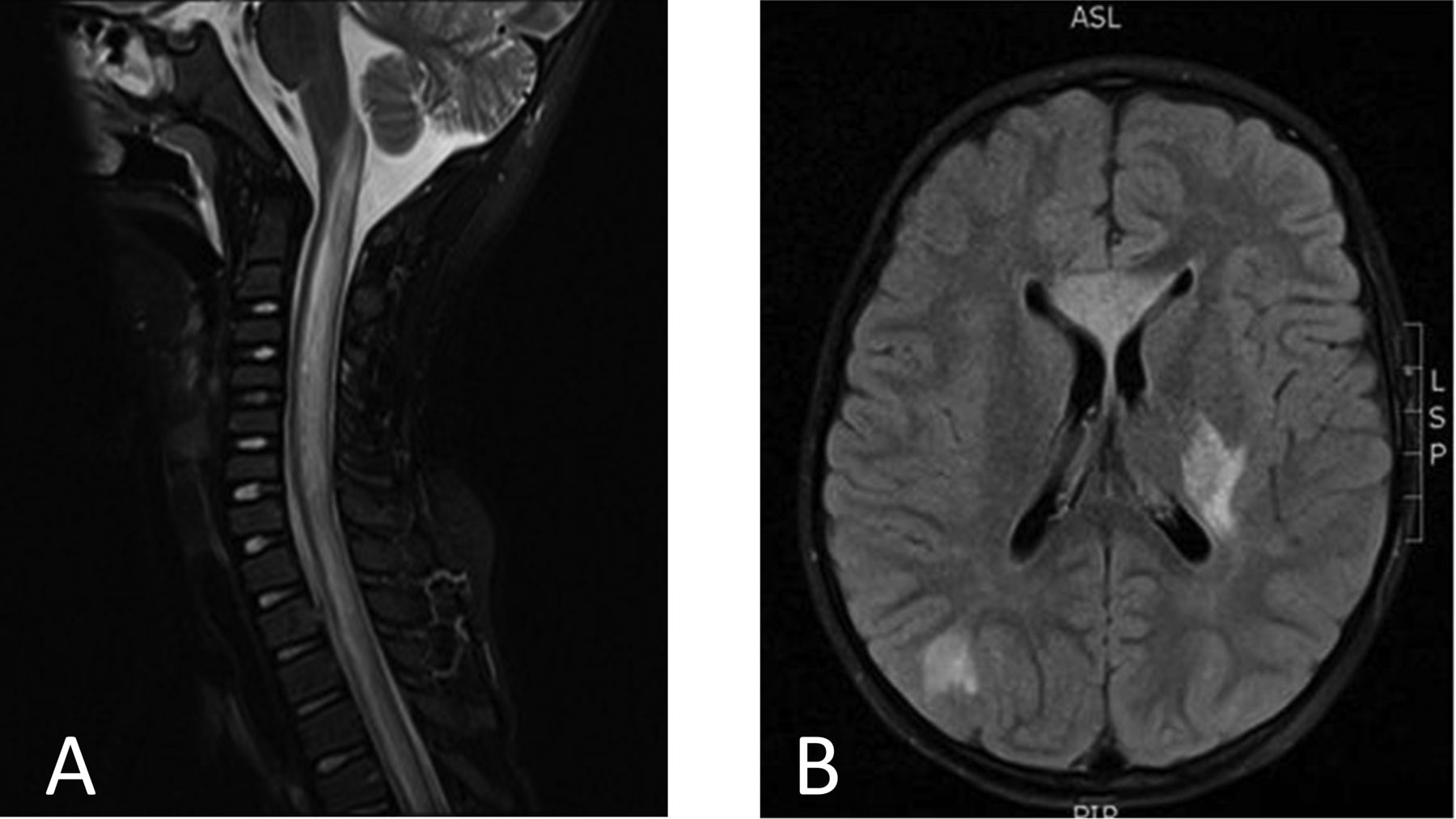
A. MRI of the cervical and thoracic spine (T2 FLAIR with contrast shown) obtained at the onset of symptoms, prior to treatment. There is a contiguous hyperintense, centrally located spinal cord lesion. B. MRI of the head (T2 FLAIR with contrast shown) obtained at the onset of symptoms, prior to treatment. There are multiple lesions of the supratentorial brain (especially involving the white matter). MRI: magnetic resonance imaging; FLAIR: fluid attenuated inversion recovery.
CSF was positive by ELISA for neuromyelitis optica autoantibody (NMO)-immunoglobulin G (IgG), as was the serum when subsequently tested. Blood was also positive for antinuclear antibody (ANA) at 1:160 (speckled) and anti-SSA (Ro) antibody. Anti-SSB (La), anti-dsDNA, anti-Sm, and anti-RNP antibodies, rheumatoid factor, and complement factor 3 (C3) and C4 were negative or normal. The patient also started receiving azathioprine (AZA) as a steroid-sparing agent.
The patient had no symptoms of SS, such as mouth or eye dryness, joint pain, or parotitis. An ophthalmological examination was normal. However, a minor salivary gland biopsy revealed > 1 lymphocytic aggregate per 4 mm2, consistent with a diagnosis of SS. Based on studies showing its efficacy in treating both SS and NMOSD1,2, rituximab (RTX) was started, after which the prednisone and AZA were discontinued. Repeat MRI demonstrated improvement and no new lesions. Three years after presentation, continued treatment with RTX every 6 months and taking no other medications, the patient displayed only subtle right-sided weakness and mild abducens and facial nerve weakness on examination.
NMOSD are rare in children and account for only 3.2%–8.5% of central nervous system (CNS) demyelinating disease under the age of 162. Discovery of a serum IgG autoantibody against the astrocytic water channel aquaporin-4 (AQP4) in this disorder has improved understanding of the range of NMOSD. While clinical NMOSD are present in 92% of adults with the anti-NMO antibody (NMO-IgG), essentially all children with the antibody showed recurrent disease along the continuum of NMOSD, including recurrent optic neuritis and recurrent longitudinally extensive transverse myelitis3. As a result, the NMOSD have been redefined clinically and NMO-IgG has been increasingly used to establish the diagnosis of an NMOSD in children4,5. With acute myelitis, positive NMO-IgG, and exclusion of alternative diagnoses, our patient met the recently proposed international criteria for NMOSD5.
Primary SS in children, though rare, has been reported6,7. SS is characterized by plasma cell and lymphocyte infiltration of the exocrine glands, classically resulting in clinical xerostomia and xerophthalmia. In children, however, SS often presents insidiously, most commonly with recurrent parotid swelling, but also with a broad spectrum of symptoms outside of the typical sicca syndrome seen in adults6,7. CNS involvement is well documented in both pediatric and adult patients with SS8. Patients with SS usually have anti-SSA and/or anti-SSB antibodies, but the presence of these antibodies may not be of diagnostic significance and may be present in completely asymptomatic individuals. Because of the lack of typical symptoms in pediatric SS, diagnosis is often difficult.
NMOSD have a high prevalence of coexistent autoimmune disorders and markers. Over 40% of adult patients with NMOSD have comorbid autoimmune diseases and over 75% have autoantibodies aside from NMOIgG: as many as 64% also have a positive ANA and 15%–38% have a positive SSA4.
Interestingly, while there are multiple reports of coexisting SS and NMOSD in adults9, to our knowledge, this has not been definitively reported in pediatrics. Several patients with NMOSD have been reported to have positive anti-SSA. While these patients may also have had SS, they either did not otherwise meet diagnostic criteria for SS (because of the lack of salivary gland biopsies)10 or specific details concerning SS diagnosis were not published4. Clinical and imaging findings possibly consistent with an NMOSD along with definitive SS have also been reported, but the diagnosis of an NMOSD was not confirmed because no NMO-IgG results were reported8,11.
An intrinsic link between SS and NMOSD has been suggested12. The human salivary glands possess aquaporin-3 and aquaporin-5 channels13; AQP4 channels have been found in rodent salivary glands14. Autoimmunity in the salivary gland, in the context of SS, may result in an immune response against AQP412. Previous studies have suggested that patients with SS with neurologic deficits should have an examination for NMOSD15. Conversely, our case suggests that patients presenting with NMOSD should be evaluated for the possibility of SS, and in addition highlights the difficulties inherent in detecting and diagnosing SS in children.








{kind=link}