

Abstract
Objective. Systemic sclerosis (SSc) is a systemic connective tissue disease caused by a genetic aberrant. The involvement of the copy number variations (CNV) in the pathogenesis of SSc is unclear. We tried to identify some CNV that are involved with the susceptibility to SSc.
Methods. A genome-wide CNV screening was performed in 20 patients with SSc. Five SSc-associated common CNV that included HLA-DRB5, HLA-DQA1, IRGM, CDC42EP3, and APOBEC3A/3B were identified from the screening and were then validated in 365 patients with SSc and 369 matched healthy controls.
Results. Three hundred forty-four CNV (140 gains and 204 losses) and 2 CNV hotspots (6q21.3 and 22q11.2) were found in the SSc genomes (covering 24.2 megabases), suggesting that CNV were ubiquitous in the SSc genome and played important roles in the pathogenesis of SSc. The high copy number of HLA-DQA1 was a significantly protective factor for SSc (OR 0.07, p = 2.99 × 10−17), while the high copy number of APOBEC3A/B was a significant risk factor (OR 3.45, p = 6.4 × 10−18), adjusted with sex and age. The risk prediction model based on genetic factors in logistic regression showed moderate prediction ability, with area under the curve = 0.80 (95% CI 0.77–0.83), which demonstrated that APOBEC3A/B and HLA-DQA1 were powerful biomarkers for SSc risk evaluation and contributed to the susceptibility to SSc.
Conclusion. CNV of HLA-DQA1 and APOBEC3A/B contribute to the susceptibility to SSc in a Chinese Han population.
- SYSTEMIC SCLEROSIS
- GENETIC PREDISPOSITION TO DISEASE
- HLA ANTIGENS
- COPY NUMBER VARIATION
- CASE-CONTROL STUDIES
Systemic sclerosis (SSc), also called scleroderma, is an immune-mediated disease characterized by extensive fibrosis of the skin and associated with various degrees of chronic inflammatory infiltration and significant microangiopathy, and changes in humoral or cellular immune system1,2. According to the degree and extent of fibrosis, there are 2 major clinical subtypes: limited cutaneous SSc (lcSSc) and diffuse cutaneous SSc (dcSSc).
Large numbers of the epidemiological characteristics were significantly different between the populations with different genetic architecture. For example, the incidence of SSc ranged from 3.7–23 per 100,000 population in different ethnic groups3,4; the prevalence of SSc for women is 4-fold to 5-fold higher than that for the male population5,6. Arnett, et al7 and Mayes, et al5 found that siblings had about a 15-fold higher risk of SSc, while first-degree relatives had about a 13-fold higher risk of SSc. In addition, analysis of SSc in twins reveals low concordance for the disease. However, high concordance for the presence of antinuclear antibodies (ANA) was observed8. This evidence shows that SSc is a complex disease caused by specific genetic and genomic variants9. Several genome-wide association followup studies10,11,12,13 and case-control studies have shown multiple susceptible single-nucleotide polymorphisms (SNP) associated with susceptibility to SSc, such as PPARG13, IRF514, transforming growth factor-β receptor15, TNIP116, STAT417, RHOB, and more. However, we demonstrated previously that even for high familial risk diseases such as thyroid cancer, a few of the significant SNP could just have limited prediction power18. Some other genetic or epigenetic variation should be discovered, such as copy number variation (CNV)19. CNV is one of most important sources of genetic structural diversity in the human genome; it can cause gene structure and accordingly gene expression change. Evidence showed that at least 8.75% to 17.7% of the variation in gene expression could be explained by CNV20. Some CNV have been demonstrated to be widely associated with susceptibility to immune-mediated diseases such as ankylosing spondylitis (AS)21, rheumatoid arthritis22,23,24, systemic lupus erythematosus25,26, and others. Some evidence has demonstrated that specific CNV were associated with SSc27.
In our present study, we analyzed potential SSc-associated CNV with Agilent array comparative genomic hybridization (aCGH) and AccuCopy CNV genotyping technologies28. Genome-wide aCGH microarrays were conducted to detect CNV in 20 patients with SSc, and then 5 candidate CNV that were suspected of being related to the pathology of SSc including HLA-DRB5, HLA-DQA1, IRGM, CDC42EP3, and APOBEC3A/3B. These CNV were validated in a large Chinese Han population.
MATERIALS AND METHODS
Patients and controls
SSc mainly includes 2 subtypes: lcSSc and dcSSc. In the discovery stage, we covered both of them in 5 male and 5 female samples so that we could get some unbiased candidate CNV associated with SSc and adjusted for sex and subtype of SSc in a Chinese Han population (4 categories with sex and subtypes) for the aCGH array screening. In the validation stage, a total of 734 subjects were enrolled, including 365 cases with SSc and 369 ethnically matched healthy controls. All patients were recruited from a multicenter study that included hospitals and outpatient clinics in Shanghai, Hebei province, Sichuan province, and Hunan province in China. Patients either met the American College of Rheumatology classification criteria for SSc29 or had at least 3 out of 5 CREST features (calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia) with sclerodactyly being mandatory30.
DNA extraction, autoantibody test, and organ involvement assessment
Peripheral blood was collected from all subjects. Genomic DNA was isolated from whole blood and stored at −30°C until used in our previous study17. The following autoantibodies were detected in patient sera: ANA, anti-DNA topoisomerase I (ATA), anticentromere (ACA), anti-U1RNP (ARA), anti-RNA polymerase 3 (anti-RNAP 3), anti-Sm, anti-SSA, anti-SSB, anti-PM-1, anti-Jo1 antibodies, and rheumatoid factor. The status of the autoantibodies was measured as binary data: positive and negative, except anti-RNAP 3, which had continuous data. In addition, autoantibody detection details could be found in our previous work6. Pulmonary fibrosis was assessed with radiograph and/or computed tomography. Organ involvement was defined as Steen and Medsger suggested31.
Genome-wide CNV analysis
As in our previous study21, Agilent SurePrint G3 Human CGH 1×1M Oligo Microarray was performed for genome-wide CNV detection and genotyping. DNA was isolated from 20 patients with SSc. For each sample, 2.2 µg input genomic DNA was restriction-digested and labeled with ULS-Cy5 and ULS-Cy3 in accordance with the manufacturer’s protocol (Agilent 2010). The labeled product was then hybridized to the array and scanned on the Agilent Microarray Scanner. The data were extracted by Agilent Feature Extraction 10.7.3.1 and analyzed by Agilent Workbench 7.0 with default variables. A human genome coordinate was conversed in hg19 uniformly in our present study.
AccuCopy technology for CNV validation
Five common CNV resulting from aCGH were validated with the AccuCopy assay28 (a multiple competitive real-time PCR) by Genesky Bio-Tech. Briefly, the genomic DNA of each subject was mixed with fluorescence-labeled specific primers (Supplementary Table 1, available from the authors on request), PCR Master Mix, and a competitive DNA with known copy number for a multiple competitive real-time PCR reaction. The PCR products were diluted and loaded onto an ABI 3730XL sequencer for quantification analysis. Raw data were analyzed by Gene Mapper 4.0, and Hg19 was used for the genome build for the genomic coordinates. The peak ratio between sample DNA and the corresponding competitive DNA (S/C) was calculated and then normalized to the median of the 4 preset 2-copy reference genes, respectively. Two normalized S/C ratios were further normalized to the median value in all samples for each reference gene and then averaged. The copy number of each target fragment was determined by the average S/C ratio × 2. Cases and controls were examined and read at the same time to minimize nonrandom errors.
Statistical analysis
In the validation stage, binary logistic regression was applied to discover association between CNV and SSc, and adjusted with sex and age to estimate the marginal effects of candidate CNV (marginal effect model). The partial effect for a specific CNV was also estimated and adjusted with other remaining CNV, sex, and age (partial effect model). In the partial effect model, for each of the 5 CNV selected for validation, the remaining 4 were adjusted so that the partial effect for each CNV could be estimated simultaneously. OR and 95% CI were calculated with the R code. Association between CNV and organ involvement was conducted with binary logistic model, while association between CNV and anti-RNAP 3 (continuous variable) was conducted with linear regression models. The individual with missing age, sex, or some other information would be omitted in the regression model. Chi-square or the Fisher’s exact test was applied for an independent test between autoantibody and organ involvement. The effect size of the CNV in subgroup analyses was conducted to certain samples with specific clinical characteristics compared with normal. R packages32 “PredictABEL”33 and “pROC”34 were applied for the receiver-operating characteristic (ROC) plot.
RESULTS
CNV discovery in patients with SSc
Genome-wide CNV analysis was conducted in 20 patients with SSc with aCGH array. The demographic characteristics of the 20 samples are shown in Supplementary Table 2 (available from the authors on request). There were 344 CNV (average 57.3, SD 8.3) found in the 20 individuals, including 140 gains and 204 losses, and they covered a 24.2-megabase of the whole genome (Figure 1). CNV hotspot was observed in the 6q21.3 region, which indicated that CNV in the HLA region might be associated with SSc. In the 22q11.2 region, a CNV hotspot was also detected with a large amount of CNV loss. After filtering the common CNV and our previously reported Chinese common CNV (1440 CNV regions)35, there were a total of 31 CNV remaining (8 gains, 23 loss; average = 5, SD 2.9), some of which may be the causal/risk CNV for SSc. There was no significant difference in the length of the common and novel CNV (Wilcoxon test, p = 0.26). There were 23 genes and correspondingly 119 RNA involved with 31 novel CNV (Supplementary Table 3, available from the authors on request). There was no significant difference between the loss/gain ratio between common and new CNV (chi-square test, p = 0.11). In our SSc CNV map, the number of deletion counts was much higher than that of duplication counts (Student t test, p < 0.004), while the size of duplication was much larger than that of deletion (Student t test, p = 0.03). These results suggested that common or novel CNV may be ubiquitous in the SSc genome and be involved in the pathogenesis of SSc.

Genome-wide aCGH analysis reveals copy number variation (CNV) in the SSc population. Significant SSc-associated CNV identified by aCGH array were noted in the human genome. Red bars represents high copy number in SSc, while green bars represent low copy number in SSc. aCGH: array comparative genomic hybridization; SSc: systemic sclerosis.
Association between CNV of APOBEC3A/3B, HLA-DQA1, and SSc susceptibility
To confirm what we found in the discovery stage, 5 common CNV regions (HLA-DRB5, HLA-DQA1, IRGM, CDC42EP3, and APOBEC3A/3B), which were found in at least 2 of the above samples, were validated in the validation stage. A total of 365 patients with SSc and 369 ethnically matched healthy individuals were enrolled in our study. There were no significant differences for the proportion of men/women in cases and controls (Supplementary Table 4, available from the authors on request). The patients with SSc included 52% with lcSSc and 41% with dcSSc, while the remaining 7% were not clearly subtyped and therefore could not be assigned to either group. The autoantibody status is shown in Table 1. Four autoantibodies were observed to be significantly differently distributed between lcSSc and dcSSc subtypes: ACA (OR 2.58, 95% CI 1.2–5.7, p = 0.024), ATA (OR 0.57, 95% CI 0.35–0.91, p = 0.025), ARA (OR 2.37, 95% CI 1.22–4.62, p = 0.014), and anti-SSB (OR 0.27, 95% CI 0.09–0.83, p = 0.016). The ARA-positive proportion in women was significantly higher than in men (OR 11.5, Fisher’s exact test, p = 0.003). Associations among other clinical characteristics are shown in Table 2 and Supplementary Table 5 (available from the authors on request).
Proportion of antibody and organ involvement. Values are n (%) unless otherwise specified.
Association between serological profile and clinical characteristics in SSc. Chi-square or 1-way ANOVA was conducted to make association analysis between categorical variables or categorical continuous variables. P value < 0.05 was considered a significant association. Corresponding OR and 95% CI are shown in Supplementary Table 5 (available from the authors on request).
The copy numbers of 5 genomic regions located in or near HLA-DRB5, HLA-DQA1, IRGM, CDC42EP3, and APOBEC3A/3B were examined in 365 patients with SSc and 369 healthy individuals (Supplementary Table 3, available from the authors on request). Among all the samples, CNV genotyping rates were greater than 80%. Both marginal effect and partial effect from the logistic regressions showed that CNV of HLA-DQA1 and APOBEC3A/B were significantly associated with the risk of SSc (Table 3). Low copy number of HLA-DQA1 was a significantly protective factor for SSc (OR 0.07, p = 2.99 × 10−17) while high APOBEC3A/B was a significant risk factor (OR 3.45, p = 6.4 × 10−18). Inconsistent conclusions were found for HLA-DRB5 and CDC42EP3 between the marginal and partial effect models. High HLA-DRB5 was significantly associated with SSc in the marginal effect model (OR 2.54, p = 7.4 × 10−21), while high CDC42EP3 was significantly associated with SSc only in the partial effect model (OR 1.33, p = 0.02). Because no significant association was found between CNV of IRGM and SSc, subgroup univariate logistic regressions were conducted to validate whether CNV of IRGM was associated with certain subtypes of SSc. Case individuals from the subtypes by the status of 10 autoimmune antibodies or organ involvement were compared with controls using binary logistic regression. However, no significant association was found between IRGM and susceptibility to any subtype of SSc (Supplementary Tables 6–11, available from the authors on request).
Association between CNV and SSc in marginal and partial logistic model.
Risk prediction models established based on sex, age, and/or CNV
Compared with the OR to display the association between genetic variants and a phenotype, disease risk prediction models are more clinically useful. Herein, we report 4 prediction models of the absolute risk for patients with SSc; these models were based on a logistic regression model.
As Figure 2 shows, model A included sex, age, and all of the 5 identified CNV. Model B included the sex, age, and copy number of HLA-DQA1 and APOBEC3A/3B, which were selected with the forward conditional stepwise method from all the covariates. Model C included sex and copy number of HLA-DQA1 and APOBEC3A/3B, because these factors remain unchanged. Model D features included SSc-susceptibility genetic factor and copy number of HLA-DQA1 and APOBEC3A/3B (Table 4). The ROC curve analysis showed that HLA-DQA1 and APOBEC3A/3B combined with sex and age had moderate prediction power [area under the curve (AUC) = 0.80, 95% CI 0.77–0.83]. Thus, the CNV of HLA-DQA1 and APOBEC3A/3B would be a potentially independent genetic susceptibility factor to SSc.
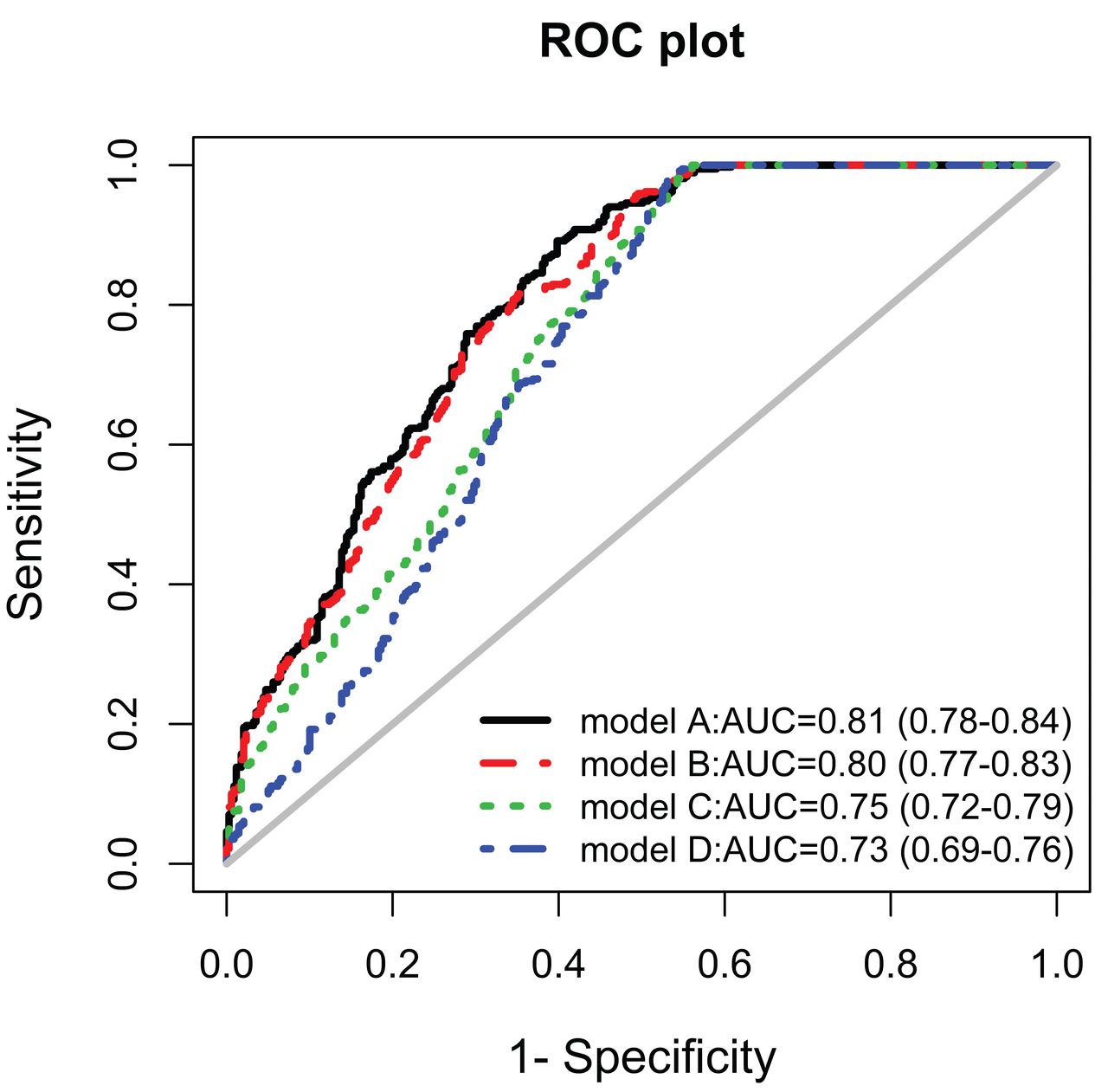
Performance of 4 prediction models based on the ROC curve. Model A included sex, age, and these CNV: HLA-DRB5, HLA-DQA1, IRGM, CDC42EP3, and APOBEC3A/3B. Model B included the sex, age, and copy number of HLA-DQA1 and APOBEC3A/3B, which were selected with a forward conditional stepwise method from all the covariates. Model C included sex and copy number of HLA-DQA1 and APOBEC3A/3B. Model D included 2 SSc-susceptibility genetic factors and the copy number of HLA-DQA1 and APOBEC3A/3B. ROC: receiver-operation characteristic; CNV: copy number variations; SSc: systemic sclerosis; AUC: area under the curve.
Distinguishing the ability of 4 models to predict risk of SSc. The outpoint corresponding to maximum of the sum of sensitivity and specificity was considered the threshold.
DISCUSSION
Our results provided evidence that the low copy number of HLA-DQA1 (OR 0.07, p = 2.99 × 10−17) and high copy number of APOBEC3A/3B (OR 3.45, p = 6.4 × 10−18) significantly contributed to the susceptibility to SSc in the Chinese Han population.
Common CNV represent an important source of genetic diversity, yet their influence on phenotypic variability and disease susceptibility remains poorly analyzed. We provided more evidence about the involvement of common CNV on complex disease, especially on immune system-related disease. Our genome-wide CNV profile in SSc and normal subjects was also supported by some previous studies, such as one finding that 22q11.2 deletion may result in variable clinical phenotypes and that 22q11.2 deletion individuals are at increased risk of a variety of autoimmune diseases36. In addition, the CNV pattern in our present study were also observed in our previous discovery in a healthy Chinese population35.
The HLA-DQA1 gene is one of the HLA complex genes encoding a protein that presents specific antigen peptides to T cell receptor to initiate immune response. Genetic allelic variation in HLA-DQA1 has been reported to be associated with SSc37. The association of CNV of HLA-DQA1 with SSc indicates dosage of HLA-DQA1 and influences the susceptibility to SSc. Additionally, CNV of HLA-DQA1 has been confirmed to be associated with many kinds of autoimmune diseases, such as AS21.
APOBEC3 are a family of DNA-editing enzymes thought to be part of the innate immune system by restricting retroviruses, mobile genetic elements such as retrotransposons, and endogenous retroviruses. APOBEC3A is an important epigenetic-related regulation factor38,39 that is highly expressed in monocytes and macrophages upon stimulation with interferon. It can activate the DNA damage response and cause cell-cycle arrest40; APOBEC3A and APOBEC3B also are potent inhibitors of long terminal repeat retrotransposon function in human cells41. In such situations, the copy number of APOBEC3A/3B will cause different reactions of the innate immune system and might have some effect on the pathogenesis of SSc.
In the prediction model section, we show the prediction performance based on CNV and some other confounders quantitatively. In addition, AUC could be used to compare with other prediction models, which were established from SNP or epidemiological factors. However, we also admit that the eventual prediction model might include all the explanatory variables such as SNP, CNV, and even some epigenetic variations in the clinical or epidemiological application in the population or hospital scenario. Moreover, the perfect prediction model should be applied 5-fold or 10-fold in cross-validation or independent dataset validation, while the sample size in our present study is limited. We would test and reevaluate our model in our future samples. We also could not detect all the significantly associated variations such as SNP, methylation, and others; thus our present study is preliminary, and we tried to identify some CNV variations associated with SSc. In the future, we could build more accurate and credible prediction models for SSc risk evaluation.
More and more aberrant CNV were found in SSc, such as FCGR3B27; therefore, genome-wide association between CNV and SSc should be studied to identify more susceptibility factors to SSc. To our knowledge, ours is the first SSc CNV map in the Chinese population, although the sample size is limited. More samples are being tested, to discover more SSc-specific or -associated CNV. In addition, we designed several probes to detect the CNV in our regions of interest. However, some CNV might be located outside the target region, which would provide underestimates for the effect size of the CNV to SSc susceptibility. Also, CNV can be limited to a single gene or include a contiguous set of genes. In the latter situation, CNV will greatly influence the human genome and so may be responsible for a substantial amount of human phenotypic variability, complex behavioral traits, and disease susceptibility. Therefore, our present study provided important evidence to identify more and more missing heritability for complex diseases based on common CNV.
Low copy of HLA-DQA1 and high copy of APOBEC3A/3B regions are significantly associated with the susceptibility to SSc.
Footnotes
Supported by research grants from the National Basic Research Program (2012CB944604), National Science Foundation of China (81270120, 81470254), International S&T Cooperation Program of China (2013DFA30870), the 111 Project (B13016), and the US National Institutes of Health (NIH) NIAID UO1, 1U01AI09090. The computations involved in this study were supported by Fudan University High-End Computing Center. Mr. Xiong was supported by Grant 1R01AR057120–01 and 1R01HL106034-01, from the NIH and the US National Heart, Lung, and Blood Institute.
- Accepted for publication January 26, 2016.








{kind=link}
{kind=link}