

Abstract
Objective. To evaluate the safety and efficacy of golimumab (GOL), a human antitumor necrosis factor antibody, in patients with active rheumatoid arthritis (RA) despite methotrexate (MTX) therapy through 5 years in the GO-FORWARD trial.
Methods. Patients with active RA despite MTX therapy were randomly assigned to receive placebo + MTX (Group 1), GOL 100 mg + placebo (Group 2), GOL 50 mg + MTX (Group 3), or GOL 100 mg + MTX (Group 4). Patients in groups 1, 2, and 3 with inadequate response could enter early escape at Week 16 to GOL 50 mg + MTX or GOL 100 mg + MTX, and all remaining Group 1 patients crossed over to GOL 50 mg + MTX at Week 24. The blind was maintained through the 52-week database lock, after which treatment adjustments were permitted. Adverse events (AE) were monitored through Week 268. Efficacy was evaluated using the American College of Rheumatology (ACR) 20/50/70 responses and a 28-joint Disease Activity Score using C-reactive protein (DAS28-CRP). Response rates at Week 256 were analyzed by an intent-to-treat analysis.
Results. A total of 444 patients were randomized, and 313 received GOL through Week 252; 301 patients completed the safety followup through Week 268. Infections were the most common type of AE; 172 patients (39.6%) had ≥ 1 serious AE. No unexpected safety signals were observed. At Week 256, ACR20/50/70 responses were achieved by 63.1%, 40.8%, and 24.1%, respectively, of all randomized patients. About 78% of all patients achieved a good or moderate DAS28-CRP response.
Conclusion. Improvements in the signs and symptoms of RA were maintained through 5 years. AE through 5 years were consistent with earlier reports of the GO-FORWARD trial; no apparent increased risk was observed over time.
The GO-FORWARD trial evaluated the safety and efficacy of subcutaneous (SC) golimumab (GOL) in patients with active rheumatoid arthritis (RA) despite methotrexate (MTX) therapy. Through Week 24, patients treated with GOL 50 mg or 100 mg + MTX had significantly greater improvements in clinical efficacy outcomes when compared with patients receiving placebo + MTX1. These improvements were maintained through 2 years, with nearly 50% of GOL + MTX-treated patients achieving disease remission as measured by the 28-joint Disease Activity Score using C-reactive protein (DAS28-CRP) at Week 1042,3. Significant improvements in health-related quality of life (HRQOL) through Week 52 were also noted for patients randomized to GOL + MTX4. Radiographic progression was minimal through 2 years in all treatment groups3,5. No unexpected safety events were observed through 2 years1,2,3. The final safety and efficacy results through 5 years of GO-FORWARD are reported herein.
MATERIALS AND METHODS
Patients and study design
The detailed GO-FORWARD eligibility criteria and study design were previously reported1. Briefly, adults with active RA (≥ 4 swollen joints, ≥ 4 tender joints) despite MTX therapy for ≥ 3 months were randomized to SC injections of placebo + MTX tablets (Group 1), GOL 100 mg + placebo tablets (Group 2), GOL 50 mg + MTX tablets (Group 3), or GOL 100 mg + MTX tablets (Group 4). Patients were stratified by investigational site. Placebo and GOL injections were administered at baseline and every 4 weeks. At Week 16, patients in groups 1–3 with < 20% improvement in swollen and tender joint counts entered double-blinded early escape (Group 1: placebo to GOL 50 mg; Group 2: began concomitant MTX while continuing GOL 100 mg; and Group 3: GOL 50 mg to 100 mg). No treatment adjustments were permitted in Group 4 at Week 16, regardless of early escape status. At Week 24, Group 1 patients still receiving placebo initiated GOL 50 mg while continuing concomitant MTX.
The longterm extension (LTE) began at Week 52 and continued through Week 268 (5 yrs). The final GOL administration was at Week 252. After Week 256, patients could transition to standard-of-care treatment, including commercially available biologics. The blind was maintained until the Week 52 database lock, after which treatment adjustments could be made at the investigator’s discretion and included a 1-time GOL dose increase to 100 mg or decrease to 50 mg (including patients who had dose-escalated to 100 mg), switching from placebo to MTX, or adjusting the MTX dose. Concomitant nonsteroidal antiinflammatory drugs (NSAID), corticosteroids, or other analgesics could also be adjusted at the investigator’s discretion.
This trial was conducted according to the Declaration of Helsinki. The protocol was approved by the institutional review board/ethics committee at each site, and all patients gave written informed consent before any study-related procedures were performed.
Evaluations
Patients were monitored through Week 268 for any adverse events (AE). AE were summarized by the actual treatment received at the time of the event. Routine laboratory analyses were performed through Week 256. Blood samples were collected prior to injections of study agents at selected visits through Week 256 for the analysis of pharmacokinetics and assessment of antibodies to GOL using a validated bridging enzyme immunoassay6.
Clinical efficacy was primarily evaluated using the American College of Rheumatology (ACR) criteria7 and DAS28-CRP scores8. Posthoc efficacy assessments included the Simplified Disease Activity Index (SDAI) and Clinical Disease Activity Index (CDAI)9. During the LTE, efficacy was evaluated every 12 weeks from weeks 52–256 with an additional assessment at Week 104.
The Health Assessment Questionnaire-Disability Index (HAQ-DI)10 was used to evaluate physical function, with a minimal clinically important difference (MCID) defined as an improvement ≥ 0.2511 and a normal physical function defined as a score ≤ 0.5. The physical and mental component summary (PCS; MCS) scores of the Medical Outcomes Study Short Form-36 (SF-36)12 were used to evaluate HRQOL, with normal scores defined as ≥ 50. The effect of disease on productivity was assessed using a visual analog scale (0–10 cm).
Radiographs of the hands and feet were obtained at weeks 52, 104, 208, and 256 during the LTE; results through Week 104 have been previously reported3,5. Data from patients with radiographs at baseline, Week 104, and ≥ 1 post-Week 104 timepoint were included in our current analysis. As previously detailed5, radiographs were scored by 2 independent readers and an adjudicator using the Sharp/van der Heijde method (SvdH)13.
Statistical analysis
Cumulative safety data were reported for all patients who received ≥ 1 GOL injection through Week 268 and summarized by the following groups: GOL 50 mg + MTX only, GOL 100 mg + placebo or MTX, and GOL 50 mg or 100 mg + placebo or MTX. Patients who received commercially available biologics (including commercial GOL) after discontinuing study GOL, but who remained in our study, had AE reported through Week 268, but these AE were excluded from the safety summaries. In addition to observed rates, the rates of serious infections, malignancies, and death were also reported as the incidence (95% CI)/100 patient-years. The standardized incidence ratios (SIR) for malignancies were determined using the Surveillance, Epidemiology, and End Results (SEER) database. Non-melanoma skin cancers were not included in the SIR analysis because these events were not reported in the SEER database.
Descriptive statistics by randomized treatment group were used to summarize the efficacy results of the LTE. For clinical efficacy measures, the protocol-specified analysis was based on observed data reported through Week 256, with no imputation for missing values. A more stringent posthoc intent-to-treat (ITT) analysis was performed for these measures and forms the basis for this report. The ITT analysis applied the following data imputation and treatment failure rules: (1) missing baseline values for continuous variables were replaced with the median value, and last observation carried forward (LOCF) methodology was applied to missing postbaseline values, and (2) patients who discontinued study agent because of unsatisfactory therapeutic effect were considered to be nonresponders. The proportions of patients were reported who achieved ≥ 20%, 50%, or 70% improvement in ACR criteria (ACR20/50/70 response), a moderate or good DAS28-CRP response8, DAS28-CRP score ≤ 3.2, an MCID in HAQ-DI score, and a HAQ-DI score ≤ 0.5. Additionally, the proportions of patients achieving remission were determined according to the following criteria: DAS28-CRP score < 2.6, SDAI ≤ 3.3, and CDAI score ≤ 2.89. SF-36 results and the effect of disease on productivity were reported using observed data only with no imputation rules for missing data.
Radiographic data at Week 256 were summarized by randomized treatment group and included all patients with radiographs at weeks 0, 104, and ≥ 1 post-Week 104 timepoint. Changes from baseline to Week 256 in total SvdH score were reported; missing postbaseline scores were replaced using the LOCF methodology. Annual rates of progression were determined for patients with total SvdH scores at baseline and Week 256 using the total SvdH score divided by RA duration at baseline and the change in total SvdH score over 5 years, respectively.
RESULTS
Patient disposition
Data for our report were collected from December 2005 to September 2012. A total of 444 patients were randomized to Group 1 (n = 133), Group 2 (n = 133), Group 3 (n = 89), and Group 4 (n = 89; Supplementary Table 1, available online at jrheum.org). Patient demographics and baseline disease characteristics were generally well balanced among the treatment groups1, and patient disposition through Week 104 has been reported1,3. A total of 106 patients (Group 1, n = 41; Group 2, n = 36; Group 3, n = 15; Group 4, n = 14) met the early escape criteria at Week 161. Among all patients, 131 (29.5%) discontinued study agent through Week 252 (Table 1); of these, 64 (14.4%) discontinued because of an AE, including worsening of RA (n = 6, 1.4%), and 25 (5.6%) discontinued because of unsatisfactory therapeutic effect. A total of 313 patients (70.5%) received GOL through Week 252, and 301 (67.8%) completed the safety followup through Week 268.
Patients who discontinued study agent through Week 252. Values are n (%) unless otherwise specified.
Types of AE
A total of 434 patients received ≥ 1 administration of GOL 50 mg or 100 mg; the mean duration of followup was 215 weeks. Of these, 105 received only the 50-mg dose, 184 received only the 100-mg dose, and 145 patients received ≥ 1 administration of each dose during the trial (Table 2). Therefore, it is difficult to make direct comparisons of the safety results between the 2 doses.
AE through Week 268. Values are n (%) unless otherwise specified.
Among all GOL-treated patients, the most common types of AE by Medical Dictionary for Regulatory Activities classification were infections/infestations (80.4%), musculoskeletal and connective tissue disorders (48.4%), and gastrointestinal disorders (46.3%) through Week 268. Common AE included upper respiratory tract infection (n = 143, 32.9%), bronchitis (n = 74, 17.1%), nasopharyngitis (n = 74, 17.1%), and cough (n = 73, 16.8%; Table 2). Forty GOL-treated patients (9.2%) reported ≥ 1 injection site reaction; none were considered to be serious or severe.
A total of 172 (39.6%) GOL-treated patients had ≥ 1 serious AE (SAE), with pneumonia and sepsis being among the most common (n = 7, 1.6% for both). The incidence [(95% CI)/100 patient-yrs] of serious infections was 4.01 (3.14–5.05; Table 2). The annual incidence/100 patient-years of serious infections among GOL-treated patients was highest during the first year of the trial (6.18, 95% CI 3.92–9.28), and then ranged from 3.26 to 1.70/100 patient-years during years 2, 3, 4, and 5 (Supplementary Table 2, available online at jrheum.org). Pneumonia, sepsis, and cellulitis were the most common serious infections, occurring in > 1.0% of patients (Table 2).
There were 4 cases of active tuberculosis (TB) throughout the trial; 2 occurred prior to Week 1043. The other 2 cases (1 pulmonary TB and 1 disseminated TB) occurred at Week 160 (1 GOL 50 mg + MTX, and 1 GOL 100 mg + MTX), both in Korea. Three opportunistic infections, all considered non-serious, occurred: candidiasis in 2 patients (1 gastrointestinal candidiasis and 1 esophageal candidiasis) and aspergilloma in 1 patient.
Twenty-seven GOL-treated patients were diagnosed with a malignancy through Week 268. The adjusted incidence/100 patient-years (95% CI) for all malignancies was 1.52 (1.00–2.20; Table 2). Twelve patients (0.67/100 patient-yrs, 95% CI 0.35–1.18) were diagnosed with non-melanoma skin cancer, and 1 patient (100 mg) was diagnosed with melanoma. Two patients (0.11/100 patient-yrs, 95% CI 0.01–0.40) were diagnosed with lymphoma; both had received GOL 100 mg + MTX. The annual rates of all malignancies and lymphoma per 100 patient-years (95% CI) for all GOL-treated patients did not indicate an increased risk with increased exposure through the 5 years (Supplementary Table 2, available online at jrheum.org).
Through Week 268, 8 deaths occurred among GOL-treated patients, corresponding to an adjusted incidence/100 patient-years (95% CI) of 0.45 (0.19–0.88; Table 2). The annual rates/100 patient-years did not indicate an increased risk over time (Supplementary Table 2, available online at jrheum.org). Four deaths (all in Group 2) occurred before Week 104 (respiratory distress, sepsis, acute hepatic failure, and cardiovascular insufficiency) and have been previously described3; 4 additional deaths occurred after Week 104. Three of these occurred among patients receiving GOL 100 mg + MTX: (1) lung cancer in a 57-year-old man with a history of chronic lung disease and cigarette smoking, (2) septic shock in a 67-year-old woman, and (3) lymphoproliferative disorder (woman, aged 59). The fourth patient, a 75-year-old man with a history of cigarette smoking who received GOL 100 mg + placebo, died of bronchial carcinoma.
Following discontinuation of study GOL injections at Week 252, 56 patients received a commercial biologic for RA (including commercial GOL) from Week 256 through Week 268. Eleven patients who received commercial GOL after Week 252 reported ≥ 1 AE through Week 268. Most of the AE were similar to those reported during receipt of study drug through Week 252. One patient had an SAE (basal cell carcinoma). This patient (woman, aged 69) had a history of basal cell carcinoma prior to enrollment, was randomized to placebo + MTX, crossed over to GOL 50 mg + MTX at Week 24, and began treatment with commercial GOL at Week 257; the patient had multiple events of basal cell carcinoma throughout the trial.
Pharmacokinetics and antibodies to GOL
Serum trough GOL concentrations were roughly dose-proportional and were generally maintained through Week 256 for patients who did not have any changes in GOL dose. Through Week 256, 429 GOL-treated patients had ≥ 1 blood sample after treatment initiation that was evaluable for antibodies to GOL. Of these patients, 32 (7.5%) tested positive for antibodies to GOL; 25 of these patients (86.2%) were positive for neutralizing antibodies. In a posthoc comparison of antibody-positive patients and antibody-negative patients, no differences were observed in the proportion of patients who discontinued because of an AE of worsening of RA (3.1% and 1.0%, respectively, p = 0.284), or because of unsatisfactory therapeutic effect (6.3% and 5.6%, respectively, p = 0.870), or had an increase in GOL dose from 50 mg to 100 mg during the LTE (25.0% and 34.0%, respectively, p = 0.331). These results should be interpreted with caution because of the relatively small number of patients who tested positive.
Of the 32 patients who were positive for antibodies to GOL, 4 (12.5%) had ≥ 1 injection site reaction (none were considered serious or severe or led to discontinuation). Forty-five (11.4%) of the 396 patients who were negative for antibodies to GOL had an injection site reaction.
Clinical efficacy and patient-reported outcomes
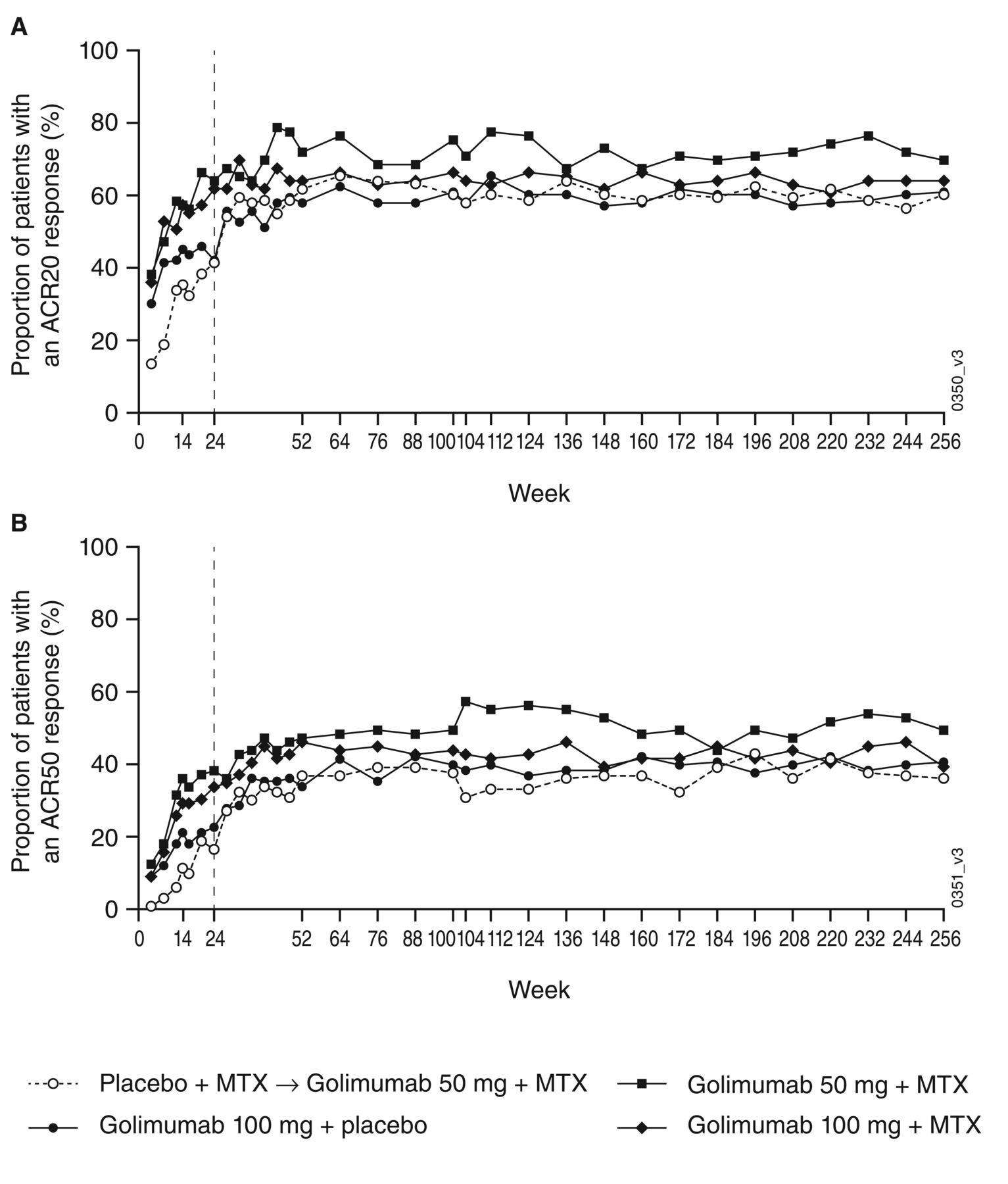
The ITT clinical efficacy results at Week 256 are shown in Table 3. Among all patients, 63.1% had an ACR20, 40.8% had an ACR50, and 24.1% had an ACR70 at Week 256, with no appreciable differences among treatment groups. ACR20 and ACR50 rates were maintained over time through Week 256 (Figure 1). At Week 256, 78.2% of all patients had a good or moderate DAS28-CRP response. About 36% of patients were in DAS28-CRP remission, while 21% met either the SDAI or CDAI remission criteria. Mean improvements from baseline to Week 256 in HAQ-DI ranged from 0.34 to 0.52, with an overall mean (SD) improvement of 0.44 (0.71). Among all patients, 61.0% had an improvement in HAQ-DI ≥ 0.25 and 36.3% achieved a normal HAQ-DI score (≤ 0.5) at Week 256 compared with 12.6% (56/444) who had a normal HAQ-DI score at baseline. The results of the protocol-specified efficacy analysis were consistent with this ITT analysis; the majority of response rates using the observed data were generally greater by about 10% or less, while some were greater by more than 10% (Supplementary Table 3, available online at jrheum.org).

The proportions of patients with an (A) ACR20 or (B) ACR50 response through Week 256 (full intent-to-treat population). ACR20/50: ≥ 20%/50% improvement in American College of Rheumatology criteria; MTX: methotrexate.
Clinical efficacy and radiographic results at Week 256. Values are n (%) unless otherwise specified.
Eighty-seven patients had an increase in GOL dose from 50 mg to 100 mg between Week 52 and Week 256 and had ≥ 12 weeks of followup. Of these, 68 patients did not have a DAS28-CRP score < 2.6 immediately prior to dose escalation; at 12 weeks after dose escalation, these patients had a mean (SD) improvement in DAS28-CRP score of 0.8 (1.2), and 14 patients (20.6%) achieved a DAS28-CRP score < 2.6. Fifty-eight patients did not have a DAS28-CRP score ≤ 3.2 immediately prior to dose escalation; at 12 weeks after dose escalation, these patients had a mean (SD) improvement in DAS28-CRP score of 1.0 (1.2), and 23 patients (39.7%) achieved a DAS28-CRP score ≤ 3.2.
Changes from baseline to Week 256 in HRQOL were generally similar among the treatment groups (Table 4). Among all patients, mean (SD) improvements from baseline to Week 256 in SF-36 PCS and MCS scores were 8.5 (10.4) and 3.9 (11.0), respectively. In addition, 24.4% and 48.7% of all patients had a normal SF-36 PCS or MCS score, respectively, compared with 1.6% and 32.1%, respectively, who had normal scores at baseline. Mean (SD) improvement in the effect of disease on productivity for all patients was 2.6 (2.9).
Improvements in health-related quality of life and the effect of disease on productivity at Week 256. Values are mean ± SD or n (%).
Among all randomized patients, concomitant oral corticosteroid use was reported by 67.3% (n = 299) of patients at baseline (mean dose: 7.4 mg/day prednisone or equivalent) and 59.2% (n = 263) at Week 256 (mean dose: 6.6 mg/day prednisone or equivalent). Concomitant NSAID use was reported by 82.7% (n = 367) of patients at baseline and 72.3% (n = 321) of patients at Week 256.
All patients received prior MTX therapy per the inclusion criteria, and concomitant MTX use through Week 52 was specified by treatment group randomization and early escape rules in the protocol. At Week 256, 74.1% (n = 329) of all randomized patients reported using concomitant MTX at a mean dose of 16.0 mg/week.
Radiographic progression
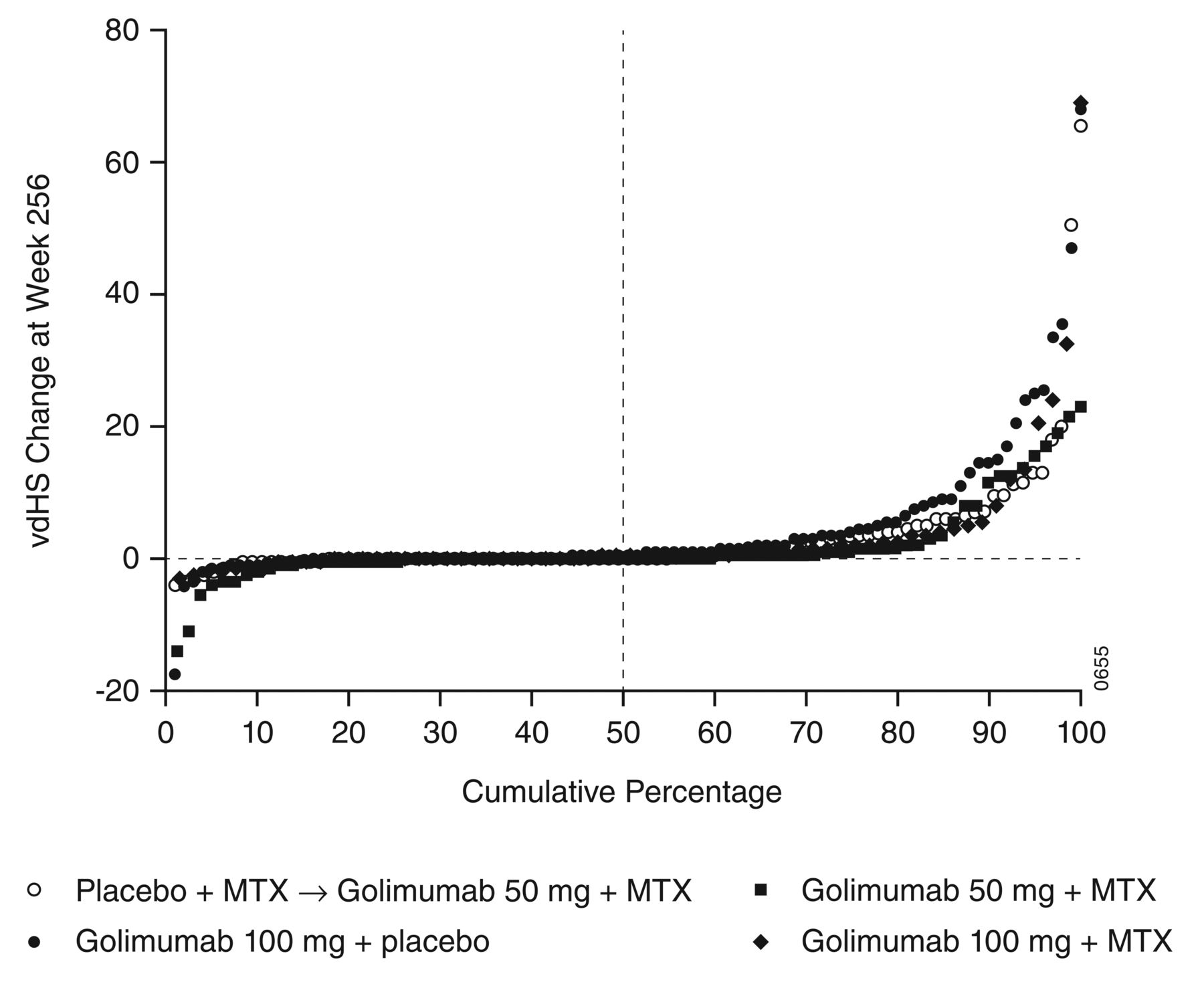
Mean changes from baseline in total SvdH scores at Week 256 for groups 1, 2, 3, and 4 were 3.17, 4.55, 1.74, and 3.29, respectively (Table 3). A probability plot of the change in total SvdH scores from baseline to Week 256 for the 4 randomized treatment groups is shown in Figure 2. Patients in groups 1 and 2, who did not receive GOL + MTX from baseline, appeared to have greater increases in total SvdH scores than did patients in groups 3 and 4. Among all patients in the final radiographic analysis, about half (n = 172, 50.9%) had a change from baseline in total SvdH score of ≤ 0 at Week 256, and 60.1% had a change in total SvdH score ≤ 0.5 (Table 3). The mean annual rate of progression through Week 256 was 0.65 for all patients (Table 3); the median annual rate of progression was 0.00 in all 4 groups (data not shown). Few patients had an annual rate of progression ≥ 5 (Table 3).

Probability plot of change in total vdHS score from baseline to Week 256. MTX: methotrexate; vdHS score: van der Heijde modification of the Sharp score.
DISCUSSION
The patients enrolled in the GO-FORWARD trial generally had longstanding RA of at least moderate activity; however, the trial population as a whole had relatively low disease activity compared with patient populations in earlier trials of patients with RA14,15. Patients treated with GOL + MTX had significantly greater improvements in disease activity, physical function, and HRQOL than did patients who received MTX monotherapy through Week 241. These improvements were maintained through 5 years as reported here.
Through 5 years, the discontinuation rate was relatively low, with about 70% of patients continuing GOL treatment through the final study drug administration at Week 252. In a previous trial with a similar patient population receiving antitumor necrosis factor (TNF) therapy, the proportion of patients continuing through 5 years was reported to be about 50%16. Safety findings through Week 268 were generally consistent with those previously reported through Week 1041,2,3, as well as with results from studies of other anti-TNF agents in patients with RA17,18,19. Safety comparisons between the 2 GOL doses are limited because of GOL dose adjustments and changes in concomitant medications that were allowed after the Week 52 database lock.
Infections were the most common type of AE, with upper respiratory infections being the most common infection among GOL-treated patients through Week 268. Infections were also the most common type of SAE, with pneumonia and sepsis being the most frequent. Four cases of active TB were reported, all in endemic areas. A total of 3 opportunistic infections (aspergilloma and candidiasis) occurred; none were considered serious. The incidence/100 patient-years of serious infections was higher with GOL ± MTX than with placebo + MTX, which is consistent with the known safety profile of anti-TNF therapies20. However, this comparison is limited by the shorter observation period and smaller population in the MTX monotherapy group. The rate of serious infections/100 patient-years was numerically highest during the first year of the trial in comparison with years 2–5. Similar trends have been noted in observational studies of patients with RA receiving anti-TNF therapies, with the greatest risk of serious infection occurring during the first 6–12 months of treatment20,21.
There were a total of 8 deaths. No apparent predominant cause of death was identified among these patients, and no increasing risk of death over time was observed through 5 years of GOL treatment.
In total, 27 GOL-treated patients were diagnosed with a malignancy, with breast cancer (n = 4) and lung cancer (n = 2) being the most common. Two patients receiving GOL 100 mg were diagnosed with lymphoma through Week 256. The corresponding SIR and 95% CI not encompassing 1 (10.06, 95% CI 1.22–36.35) suggest a possible increased risk of lymphoma with this GOL dose. Previous research has shown that patients with RA appear to be at increased risk of developing lymphoma22, with patients with more severe disease having the highest risk23,24. The involvement of RA therapies in this relationship is unclear; however, a large registry analysis25 did not find an elevated lymphoma risk with increasing duration of anti-TNF therapy in patients with RA. Of note, in our trial, the annual incidences of malignancies and lymphoma per 100 patient-years did not appear to increase through 5 years.
Serum trough GOL concentrations were roughly dose-proportional and were maintained through Week 256. Less than 8% of patients were positive for antibodies to GOL through Week 256, which was consistent with other phase III trials of subcutaneous GOL in patients with RA26,27, ankylosing spondylitis28, and psoriatic arthritis29. Few patients who tested positive for antibodies had an injection site reaction. There were no apparent differences between antibody-positive patients and antibody-negative patients in the proportion of patients who discontinued because of an AE of worsening RA or because of unsatisfactory therapeutic effect, or the proportion of patients with a GOL dose escalation from 50 mg to 100 mg during the LTE. The anti-GOL antibody results from our study should not be compared with trials of other anti-TNF agents because the methods used for determining the presence of antidrug antibodies and sampling times during our trial may have large influences on the results.
Clinical response was sustained through 5 years, with 63.1% of patients having an ACR20 response at Week 256 and 78.2% having a good or moderate DAS28-CRP response. Improvements in physical function were also maintained through 5 years with 61% of patients having a clinically important improvement in HAQ-DI score at Week 256. Radiographic progression was minimal through 5 years, and at Week 256, about half of all patients had a change in total SvdH score ≤ 0.
From baseline to Week 256, the proportion of patients receiving concomitant oral corticosteroids and the mean dose decreased, as did the proportion of patients receiving concomitant NSAID. These findings may suggest that the use of these concomitant medications may be reduced with GOL treatment. It should be noted that the use of these medications was solely at the discretion of the investigator.
Interpretation of the longterm results of the GO-FORWARD is limited by the inclusion of patients with few comorbidities at baseline, which may affect the AE reported during the trial, as well as selection bias over time and the changes allowed to patients’ GOL dose and concomitant medications. Additionally, the trial population consisted of patients who had active disease despite prior treatment with MTX, which may limit the generalizability of our results to other RA patient populations, such as those with early disease or those previously treated with other anti-TNF therapies.
ONLINE SUPPLEMENT
Supplementary data for this article are available online at jrheum.org.
Acknowledgment
The authors thank Rebecca Clemente, PhD, and Mary Whitman, PhD, of Janssen Scientific Affairs LLC for writing support.
Footnotes
Funded by Janssen Research & Development LLC, a wholly owned subsidiary of Johnson & Johnson, and by Merck/Schering-Plough. E.C. Keystone has received research grants, consulting fees, and honoraria from Janssen. M.C. Genovese has received research grants and consulting fees from Janssen Research & Development LLC. S. Hall and S.C. Bae have received consulting fees from Janssen Research & Development LLC. C. Han and T.A. Gathany are employees of Janssen Global Services LLC, and own stock in Johnson & Johnson. S. Xu, Y. Zhou, J.H. Leu, and E.C. Hsia are employees of Janssen Research & Development LLC, and own stock in Johnson & Johnson.
- Accepted for publication October 15, 2015.








{kind=link}
{kind=link}