

Abstract
Objective. To evaluate subcutaneous SBI-087 to treat rheumatoid arthritis (RA).
Methods. A total of 210 adult patients with active RA were randomized to receive either 200 mg SBI-087 or placebo (Pbo), according to one of these patterns: SBI/Pbo/Pbo (SBI on Day 1), SBI/SBI/Pbo (SBI days 1 and 15), SBI/Pbo/SBI (SBI days 1 and 84), SBI/SBI/SBI (SBI days 1, 15, and 84), or Pbo/Pbo/Pbo (Pbo all 3 days). All patients were seropositive and taking background methotrexate. The primary endpoint was proportion of patients achieving 20% improvement from baseline at Week 16 by American College of Rheumatology criteria (ACR20). Other outcomes included 28-joint Disease Activity Score (DAS28)–C-reactive protein (CRP), physician’s and patient’s global assessments of disease activity (PGA and PtGA, respectively) and Health Assessment Questionnaire–Disability Index (HAQ-DI). Peripheral CD19+ B cells were measured by high-sensitivity flow cytometer. Statistical significance was set at 2-sided α 0.10 level.
Results. The SBI/SBI/SBI group demonstrated significant improvement in ACR20 and DAS28-CRP from Week 8 onward, sustained improvement in CRP levels from Week 12 onward, and significant improvements in PGA and PtGA in weeks 16 through 24, and in HAQ-DI at Week 24. The SBI/Pbo/Pbo and SBI/SBI/Pbo groups did not meet the primary endpoint but demonstrated improvements in several secondary endpoints. All treatment groups exhibited depletion of peripheral CD19+ B cells throughout the study. Overall, 61.5% of patients receiving SBI-087 and 55.0% of patients receiving Pbo reported adverse events.
Conclusion. SBI-087 effectively depleted peripheral CD20 B cells and was well tolerated. Improvements were consistently observed in the SBI/SBI/SBI group for the majority of efficacy and quality-of-life outcomes.
Rheumatoid arthritis (RA) is a chronic, systemic, autoimmune, inflammatory disease of unknown etiology that affects about 1% of the population worldwide1,2. It is a painful condition characterized by changes in synovial tissue and symmetric inflammation of small peripheral joints, and when left untreated can progress to cartilage and bone damage2,3.
Current options in the treatment of RA include methotrexate (MTX), leflunomide (LEF), sulfasalazine, kinase inhibitors (tofacitinib)4, and biologic agents such as tumor necrosis factor-α (TNF-α) antagonists [adalimumab (ADA), certolizumab pegol (CZP), etanercept (ETN), golimumab (GOL), infliximab (IFX)], B cell–depleting agents [rituximab (RTX)], and agents targeting T cells [abatacept (ABA)] and interleukin 6 receptors [tocilizumab (TCZ)]1,5. Despite the spectrum of agents available, there is as yet no cure for RA, and up to one-third of patients remain refractory to treatment, signifying a substantial unmet medical need6.
Although the underlying etiology of RA is not fully understood, there is considerable evidence that B cells play a significant role in disease pathology, as demonstrated by the efficacy of B cell depletion for management of RA in patients who do not respond to disease-modifying antirheumatic drugs (DMARD) or anti-TNF therapies1,7,8,9,10,11.
SBI-087 is a humanized small modular immunopharmaceutical (SMIP) biologic agent, and like RTX, targets the CD20 antigen expressed on B cells12. SMIP biologics are single-chain polypeptides, smaller than monoclonal antibodies, comprising a binding domain, a hinge domain, and an effector domain13. In vitro, SBI-087 binds with high affinity to CD20 on human B cells, mediates potent complement-mediated cellular cytotoxicity, and potentiates Fc-mediated antibody-dependent cellular cytotoxicity in the presence of effector cells. These in vitro observations were translatable to in vivo. Following SBI-087 administration in cynomolgus monkeys, profound B cell depletion was observed in serum, lymph nodes, and bone marrow14. The first human studies examined single intravenous (up to 2.0 mg/kg) and subcutaneous (SC; up to 300 mg) injections of SBI-087 in patients with RA and systemic lupus erythematosus (SLE)15. Unanticipated reactions following SC administration of SBI-087 included fever, chills, and malaise, seen on the day of dosing for the lowest-dose cohorts in both indications. These were abrogated in subsequent cohorts by a pre/post-dose treatment regimen consisting of oral corticosteroids, acetaminophen, and an antihistamine. Overall, SBI-087 was considered well tolerated when given SC with pre- and post-dose treatment regimens15.
The primary objective of our current study was to evaluate the clinical efficacy and safety of 4 SBI-087 SC dosing regimens compared with placebo (Pbo) in patients with active RA who were seropositive for rheumatoid factor (RF) or anticyclic citrullinated peptide antibodies (anti-CCP) and taking MTX.
MATERIALS AND METHODS
Study design and treatments
This was a phase II, multicenter, randomized, Pbo-controlled, double-blind, parallel-group study conducted in 12 countries and registered with ClinicalTrials.gov, NCT01008852. The study was conducted in 2 stages. The first was a double-blind treatment stage (screening to Week 24), which included the primary and secondary efficacy and safety analyses. The second was a longterm followup stage in which patients were followed to monitor repletion of CD19+ B cell levels and safety. The results of the double-blind treatment stage are presented herein.
Patients were stratified based on having received prior anti-TNF treatment and into specific geographic regions. Four dosing regimens using 200 mg SC injections were tested. Patients were randomized equally to one of 5 treatment groups: Pbo and 4 treatment regimens with SBI-087 (Supplementary Figure 1, available online at jrheum.org). Patients received either SBI-087 200 mg or placebo SC at baseline, Day 15, and Day 84. The rationale for the regimens was that 1 regimen assessed the minimum dosing frequency (Day 1 only); a second tested the maximum dose frequency (Day 1, Day 15, and Day 84); the other 2 evaluated 2 doses, with the second being administered at different timepoints (Day 15 vs Day 84) in anticipation that the timing of the second dose could affect the degree of B cell depletion, and ultimately the clinical outcome. The 200-mg dose was chosen based on SBI-087 B-cell interaction modeling and findings in the first human study in patients with RA15. The timings of the dose administrations were also model-derived and suggested that B cell repletion may ensue shortly before Day 84, resulting in the final dose being administered on Day 84.
Owing to the reactions seen in the phase I studies, all doses of study medication were administered with 40 mg of prednisone or equivalent, 500 mg of acetaminophen, and an antihistamine given within 2 h pre-dose, and 20 mg of prednisone or equivalent and 500 mg of acetaminophen 4 h after dosing. The study protocol was reviewed and approved by an independent ethics committee at each institution prior to initiation. The study was conducted in compliance with the ethical principles of the Declaration of Helsinki and the International Conference on Harmonization Good Clinical Practice Guidelines. All patients provided written informed consent.
Patients
Patients, aged ≥ 18 years, were eligible if they had a confirmed diagnosis of active RA for at least 6 months [≥ 5 swollen joints and ≥ 5 tender joints, C-reactive protein (CRP) ≥ 7 mg/l or erythrocyte sedimentation rate (ESR) ≥ 28 mm/h], diagnosed according to the American Rheumatism Association revised criteria of 198716. They also had to be in American College of Rheumatology (ACR) functional class I to III, seropositive for RF and/or anti-CCP, and taking MTX (up to 25 mg/week) for ≥ 12 weeks with ≥ 8 weeks of stable dose and route of administration of the drug. Patients were excluded from the study if they had any of the following conditions: pregnancy, severe heart failure, tuberculosis (active or history of), other rheumatic diseases, cancer (active or history of), immunodeficiency disease, history of drug or alcohol abuse, or any medical condition or infection that could be worsened if patients participated in the study, in the opinion of the investigator. Patients were also excluded if they had previously received ABA, ADA, CZP, GOL, IFX, RTX, or TCZ ≤ 10 weeks of baseline visit, ETN ≤ 4 weeks of baseline visit, live vaccine ≤ 8 weeks prior to screening, investigational drugs ≤ 24 weeks prior to the baseline visit, other DMARD besides MTX, LEF ≤ 12 weeks prior to the baseline visit, or had known hypersensitivity to biopharmaceutical proteins.
Assessments
The primary efficacy endpoint was the proportion of patients who achieved 20% improvement from baseline to Week 16 according to ACR criteria (ACR20)17. Secondary efficacy assessments included ACR50 (50% improvement from baseline), ACR70 (70% improvement from baseline), 28-joint Disease Activity Score (DAS28)-CRP, swollen joint count of 28 joints (SJC28), tender joint count of 28 joints (TJC28), and CRP levels. Health-related quality of life (HRQOL) outcomes included assessment of pain, general health, health assessment questionnaire disability index (HAQ-DI), Functional Assessment of Chronic Illness Therapy-Fatigue, morning stiffness, Medical Outcomes Study Short Form-36 survey (SF-36), patient’s global assessment of disease activity (PtGA), and physician’s global assessment of disease activity (PGA).
The primary pharmacodynamic evaluation was the analysis of peripheral B cell counts. B cells were obtained at baseline and weeks 2, 4, 8, 12, 16, and 24, and were quantified using a high-sensitivity flow cytometry assay with a lower limit of quantification of 0.3 cells/μl. Safety evaluations included adverse events (AE), serious AE (SAE), safety laboratory tests, physical examinations, and vital signs. Patients were evaluated for safety at every visit and with a followup call within 24 hours of treatment administration.
Statistical analyses
For the primary efficacy endpoint, a sample size of 40 subjects per group provided ≥ 80% power to detect a 30% ACR20 difference between each SBI-087 treatment group and placebo at Week 16 using a 2-sided chi-squared test, with a significance level of 0.1. There were no adjustments made for multiple comparisons. Efficacy and safety analyses were performed on the modified intent-to-treat population that included all randomized patients who received treatment.
For assessments during the double-blind treatment stage, changes from baseline in efficacy outcomes were evaluated using ANCOVA, with treatment, prior anti-TNF use, and specific geographic region as factors and baseline as covariate. Each of the comparisons of SBI-087 versus placebo was performed using a 2-sided Cochran-Mantel-Haenszel test, stratified by prior anti-TNF treatment and specific geographic region. Missing efficacy data were imputed using the last observation carried forward (LOCF) method. Descriptive summaries are provided for continuous and ordinal variables.
RESULTS
Patient disposition and demographics
A total of 210 patients were randomized to the 5 treatment groups (SBI/Pbo/Pbo, n = 43; SBI/SBI/Pbo, n = 42; SBI/Pbo/SBI, n = 43; SBI/SBI/SBI, n = 41; Pbo/Pbo/Pbo, n = 41), of whom 209 received either SBI-087 or placebo; 1 patient in the Pbo/Pbo/Pbo group did not receive treatment (Figure 1). A total of 37 patients discontinued from the study. The major reasons for discontinuation were AE and lack of efficacy. The baseline characteristics were generally similar across all treatment arms (Table 1).

Patient disposition. One patient in the Pbo/Pbo/Pbo group did not receive study medication. SBI/Pbo/Pbo = 200 mg SBI-087 on Day 1. SBI/SBI/Pbo = 200 mg SBI-087 on days 1 and 5. SBI/Pbo/SBI = 200 mg SBI-087 on Day 1 and on Week 12. SBI/SBI/SBI = 200 mg SBI-087 on days 1 and 15, and on Week 12. Pbo/Pbo/Pbo = placebo.
Baseline demographics and disease characteristics. Data are given as mean (SD) unless otherwise indicated.
Efficacy
Among the treatment regimens, the onset in differentiating from placebo for ACR20 occurred as early as Week 8. Compared with patients in the Pbo/Pbo/Pbo group, statistically significant improvement in the proportion of patients who achieved ACR20 at Week 16 (p = 0.046) and Week 24 (p = 0.053; Figure 2A) was observed for the SBI/SBI/SBI group. None of the other treatment groups demonstrated significant improvement in the proportion of patients who achieved ACR20 at Week 16 compared with placebo. A statistically significant greater proportion of patients in the SBI/Pbo/Pbo (p = 0.094), SBI/SBI/Pbo (p = 0.005), and SBI/Pbo/SBI (p = 0.060) groups achieved ACR50 at Week 12 compared with the Pbo/Pbo/Pbo group (Figure 2B). However, this improvement was not sustained. Compared with patients in the Pbo/Pbo/Pbo group, a statistically significant greater proportion of patients in the SBI/SBI/SBI (p = 0.020) and SBI/Pbo/SBI (p = 0.033) groups achieved ACR70 at Week 24 (Figure 2C).
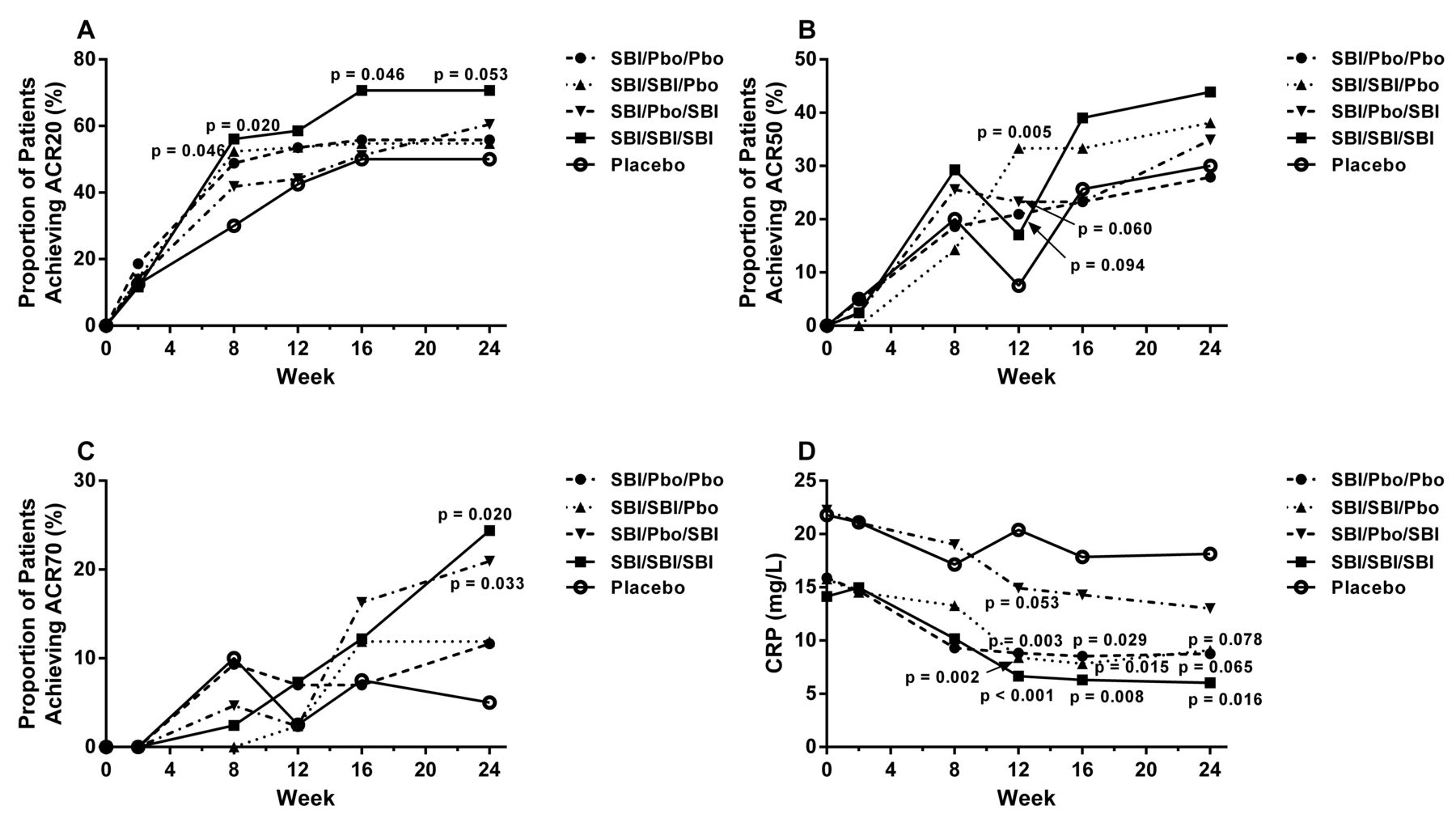
Summary of efficacy responses over time. A. ACR20. B. ACR50. C. ACR70. D. CRP levels. ACR20/50/70: American College of Rheumatology percent of improvement from baseline; CRP: C-reactive protein; SBI: SBI-087; Pbo: placebo.
All SBI-087–treated groups, except the SBI/Pbo/SBI group, exhibited sustained and statistically significant decrease from baseline in CRP levels compared with the Pbo/Pbo/Pbo group from Week 12 through to Week 24 (Figure 2D). The SBI/Pbo/SBI group exhibited statistically significant decrease from baseline in CRP levels compared with the Pbo/Pbo/Pbo group at Week 12 (p = 0.053), but this response was not sustained through the double-blind phase of the trial (Figure 2D).
Sustained improvements from Week 16 through Week 24 that were statistically significant compared with the Pbo/Pbo/Pbo group were observed in the SBI/SBI/SBI group for general health (p = 0.008, and p = 0.067, respectively) and PtGA (p = 0.070 and p = 0.018, respectively; Table 2). In addition, statistically significant improvements compared with the Pbo/Pbo/Pbo group were observed in the SBI/SBI/SBI group at Week 16 for pain (p = 0.035) and morning stiffness (p = 0.026; Table 2).
Adjusted mean change [standard error of the mean (SEM)] from baseline in secondary endpoints.
For the longitudinal efficacy score DAS28-CRP, significant changes from baseline were seen by Week 16 (−2.05; p = 0.027) and also at Week 24 (−2.14; p = 0.005) for the SBI/SBI/SBI-treated group (Table 2) compared with the Pbo/Pbo/Pbo group by Week 16 (−1.45) and at Week 24 (−1.34). Although there was no statistically significant difference observed between the SBI/SBI/SBI and Pbo/Pbo/Pbo groups at either Week 16 or Week 24 for SJC28, a statistically significant difference (p = 0.007) between these groups was observed at Week 24 for TJC28 (Table 2). The SBI/SBI/SBI group also exhibited statistically significant difference compared with the Pbo/Pbo/Pbo group in PGA at both Week 16 (p = 0.081) and Week 24 (p = 0.083; Table 2).
Pharmacodynamics
Following SBI-087 administration, CD19+ B cell depletion was observed for the first 24 weeks across all SBI-087 treatment groups (Figure 3). The extent and duration of the B cell depletion profiles closely followed the dosing regimen used for each treatment group. The maximum and least duration of B cell depletion were observed in the SBI/SBI/SBI and SBI/Pbo/Pbo treatment groups, respectively. At the individual level, all the patients receiving SBI-087 showed some degree of modulation in the B cell concentrations. Patients in the Pbo/Pbo/Pbo group did not experience B cell depletion. Only 1 patient who received active treatment (SBI/Pbo/SBI) was positive for antibodies to SBI-087 post baseline; this sample was also positive for neutralizing antibodies. There was no effect of the neutralizing antibodies on the pharmacokinetics, CD19+ B cell depletion, or safety for this patient.
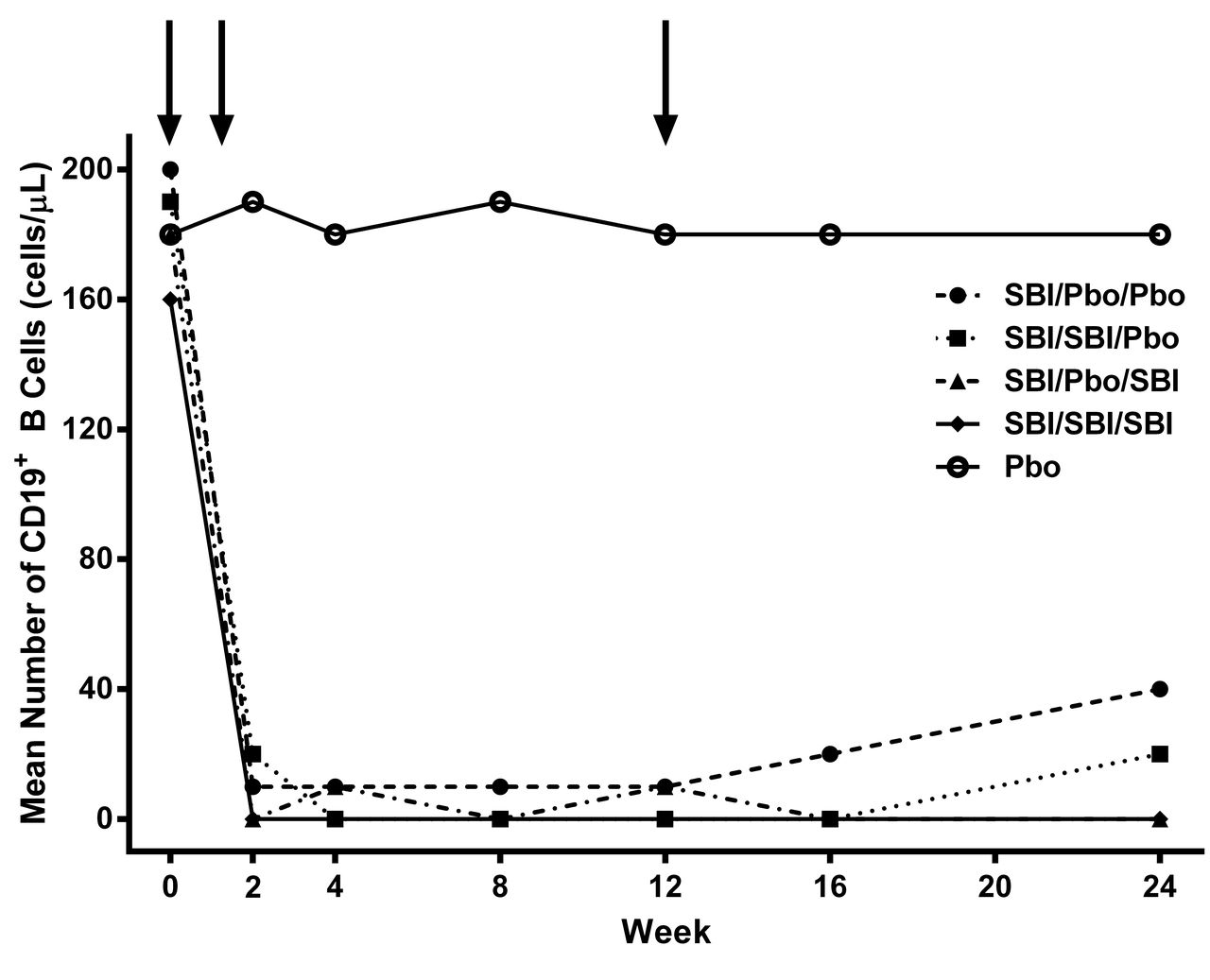
CD19+ B cell profiles after treatment. Arrows indicate treatment times. SBI: SBI-087; Pbo: placebo.
HRQOL
Statistically significant improvements compared with the Pbo/Pbo/Pbo group were observed in the SBI/SBI/SBI group at Week 24 for HAQ-DI (p = 0.055) and the SF-36 physical component score (p = 0.095; Table 2). There were few statistically significant differences among the responses of patients in any of the SBI-087 treatment groups and those receiving placebo for any of the other HRQOL measures at either Week 16 or Week 24 (Table 2).
Safety
Overall, during the double-blind phase, 104/169 (61.5%) patients in the SBI-087 treatment groups and 22/40 (55.0%) patients in the Pbo/Pbo/Pbo group experienced at least 1 AE (Table 3). There were no clinically relevant differences between the groups treated with SBI-087 and those receiving placebo. The most commonly reported AE (> 5% of patients in any treatment group) during the double-blind phase based on Medical Dictionary for Regulatory Activities coding were bronchitis, diarrhea, headache, hypertension, influenza, leukopenia, nasopharyngitis, upper respiratory tract infection, and urinary tract infection. During the double-blind phase, SAE were reported for 11 patients (6.5%) treated with SBI-087 and 3 patients (7.5%) receiving placebo. Of the SAE, 2 were serious infections (necrotizing pneumonia and wound infection). There was 1 death in the SBI/Pbo/SBI group; the patient had preexisting chronic obstructive pulmonary disease and died from severe respiratory failure and necrotizing pneumonia — only the necrotizing pneumonia was considered related to treatment by the investigator. A total of 15 patients permanently discontinued treatment because of AE (Table 3). Decreases in immunoglobulins (IgA, IgG, and IgM) were seen through Week 24 for all treatment groups; however, the decreases were not clinically significant and were not associated with infections.
Treatment-emergent adverse events during the double-blind phase. Data are given as n (%).
DISCUSSION
SBI-087 is a SMIP that targets CD20 on B cells and was investigated as a potential treatment for patients with RA. In vitro and in vivo studies showed that SBI-087 had antibody-dependent cellular cytotoxicity and complement-dependent cellular cytotoxicity activity and resulted in profound B cell depletion14. The profound B cell depletion was confirmed in 2 phase I studies in patients with mild RA and SLE who were administered SBI-087 intravenously or SC15. The phase I study in RA showed that SC doses of ≥ 100 mg showed depletion to < 0.3 cells/μl. This phase II study was designed to determine which treatment regimen would provide sustained B cell depletion, show efficacy, and be safe.
Treatment with SBI-087 at all 3 timepoints resulted in statistically significant differences compared with Pbo/Pbo/Pbo for ACR20 at weeks 16 and 24, and ACR70 at Week 24. When efficacy was measured as DAS28-CRP, patients in the SBI/SBI/SBI group exhibited improvement from Week 8 through Week 24 compared with those receiving Pbo/Pbo/Pbo. When response was measured as a decrease from baseline in CRP levels, patients in the SBI/Pbo/Pbo, SBI/SBI/Pbo, and SBI/SBI/SBI groups showed statistically significant differences compared with those in the Pbo/Pbo/Pbo group from Week 12 through to Week 24. Throughout the study, patients in the SBI/SBI/SBI group consistently exhibited improvement in each of the efficacy variables evaluated whereas results were not as consistent for patients in the other 3 arms receiving SBI-087.
In this phase II study, B cell levels were followed until they reached the lower limit of quantitation (0.3 cells/μl). The B cell depletion profiles closely followed the regimen administered, providing confidence in the targeting mechanism of SBI-087. The B cell baseline levels were similar across the different treatment groups, and the rate of depletion seemed similar across the groups receiving SBI-087. The duration of depletion, however, was most profound in the SBI/SBI/SBI group, followed by the SBI/SBI/Pbo group. These data suggest that frequent administration of SBI-087 about every 12 weeks may be needed to maintain B cell depletion.
A close relationship in continued B cell depletion and continued decrease in CRP from baseline as reflected in the shape of the time profiles of B cell depletion and CRP was observed. For the SBI/SBI/SBI group, both B cell depletion and decrease in CRP occurred up to Week 24. Similarly for SBI/Pbo/Pbo, B cell levels started to replete around Week 8 and the decrease in CRP levels plateaued around Week 8 as well. Exactly the same trends were observed for DAS28-CRP, which was probably driven by the contribution of CRP to DAS28-CRP scores.
As with the efficacy variables, compared with patients in the Pbo/Pbo/Pbo group, statistically significant improvements from baseline were observed for the SBI/SBI/SBI group in PtGA and general health from Week 16 through Week 24 and in HAQ-DI at Week 24. Patients in this group also exhibited significant improvements in pain and morning stiffness at Week 16, but the improvements were not sustained. These data support the idea that frequent administration of SBI-087 may be needed for efficacy and HRQOL outcomes. There were no statistically significant differences in number or severity of AE between the patients treated with SBI-087 at any dose and those receiving placebo.
These data indicate that SBI-087 was well tolerated but demonstrated efficacy only with the SBI/SBI/SBI regimen for the majority of efficacy and HRQOL outcome measures by Week 24. Although the other dosing regimens resulted in depletion of CD19+ B cells, they did not consistently demonstrate efficacy or improve HRQOL. The high placebo response in all categories of measurement may reflect the pulse dosing of prednisone in all patients at each dosing schedule. It is expected that SBI-087 will be delivered with a 40-mg dose of oral prednisone prior to administration of the injection and 20 mg of oral prednisone after dosing during phase III studies. However, it is possible that future studies could be conducted to determine whether the oral prednisone dose could be lowered or perhaps eliminated after the first few doses in subjects who did not initially develop symptoms. This would reduce potential AE that could be seen with regular or longterm dosing of SBI-087 and oral prednisone. The lack of consistency of the data raises concerns about the utility of SBI-087 as a therapeutic agent to treat RA, although alternative dosing regimens could be investigated to determine its efficacy and safety.
ONLINE SUPPLEMENT
Supplementary data for this article are available online at jrheum.org.
Acknowledgment
Medical writing support was provided by Mukund Nori, PhD, MBA, CMPP, of Engage Scientific Solutions and funded by Pfizer, New York, New York, USA.
Footnotes
Funded by Pfizer. Drs. Diehl, Bhattacharya, Peeva, and Menon are employees of Pfizer and own stock in Pfizer. Dr. Gourley is an employee of Janssen.
- Accepted for publication August 25, 2016.








{kind=link}
{kind=link}
{kind=link}