

Abstract
Objective. Individual dermatomyositis (DM)-associated autoantibodies are associated with distinct clinical phenotypes. This study was undertaken to explore the association of these autoantibodies with specific muscle biopsy features.
Methods. DM subjects with a muscle biopsy reviewed at Johns Hopkins had sera screened for autoantibodies recognizing Mi-2, transcriptional intermediary factor 1-γ (TIF1-γ), NXP2, MDA5, Ro52, PM-Scl, and Jo1. We also included anti-Jo1–positive patients with polymyositis (PM) who had a biopsy read at Johns Hopkins. Analyzed histological features included perifascicular atrophy, perivascular inflammation, mitochondrial dysfunction, primary inflammation, and myofiber necrosis. Duration of disease, biopsy location, and treatment at biopsy were also analyzed.
Results. We studied 91 DM and 7 anti-Jo1–positive patients with PM. In univariate analyses, TIF1-γ+ patients had more mitochondrial dysfunction (47% vs 18%; p = 0.05), NXP2+ patients had less primary inflammation (0% vs 28%; p = 0.01), Mi-2+ patients had more primary inflammation (50% vs 19%; p = 0.03), and PM-Scl+ patients had more primary inflammation (67% vs 18%; p = 0.004) than those who were negative for each autoantibody. Although reliability was limited because of small sample numbers, multivariate analysis confirmed that TIF1-γ+ patients had more mitochondrial dysfunction [prevalence ratio (PR) 2.6, 95% CI 1.0–6.5, p = 0.05] and PM-Scl+ patients had more primary inflammation (PR 5.2, 95% CI 2.0–13.4; p = 0.001) independent of disease duration at biopsy, biopsy site, and treatment at biopsy. No differences in muscle biopsy features were noted between anti-Jo1–positive patients diagnosed with DM and PM.
Conclusion. The prevalence of different histological features varies according to autoantibody status in DM. Muscle biopsy features are similar in anti-Jo1 patients with and without a rash.
Dermatomyositis (DM) and polymyositis (PM) are acquired autoimmune myopathies characterized by symmetric proximal muscle weakness, elevated muscle enzymes, and inflammatory infiltrates on muscle biopsy1. The classic diagnostic criteria of Bohan and Peter distinguished DM from PM based exclusively on the presence or absence of characteristic DM rashes1. However, in the nearly 40 years since those criteria were published, muscle biopsy features characteristic of both DM and PM have been described. According to more modern classification systems, the pathognomonic histologic feature of DM is perifascicular atrophy, while PM is characterized by lymphocytes surrounding and invading non-necrotic muscle fibers (i.e., primary inflammation)2,3,4,5. Interestingly, histological evidence of mitochondrial dysfunction has been reported as a characteristic feature in some patients with DM6. In addition, it is now recognized that some patients with autoimmune myopathy have a predominantly necrotizing myopathy with minimal inflammatory cell infiltrates and no perifascicular atrophy. These patients are now categorized histologically as having immune-mediated necrotizing myopathy (IMNM) or necrotizing autoimmune myopathy (NAM) rather than DM or PM2,7.
Another major advance in our understanding of the autoimmune myopathies is the recognition that distinct autoantibodies are associated with unique clinical phenotypes. Examples include the following: (1) patients with one of the antisynthetase antibodies (e.g., anti-Jo1, anti-PL7, and anti-PL12) have a syndrome (the antisynthetase syndrome) that includes myositis, interstitial lung disease, a nonerosive arthritis, mechanic’s hands, and/or Raynaud phenomenon8; (2) among patients with cancer-associated DM, 83% (24/29) have antibodies against either transcriptional intermediary factor 1-γ (TIF1-γ) or NXP29; (3) anti-NXP2–positive patients frequently develop calcinosis10; and (4) anti-MDA5–positive patients have prominent skin ulcerations and distinctive palmar papules11,12.
It has been shown that anti-signal recognition particle (anti-SRP) and anti–3-hydroxy-3-methylglutaryl-CoA (Coenzyme A) reductase (anti-HMGCR) autoantibodies are associated with necrotizing muscle biopsies, and patients with these serologic profiles have IMNM/NAM7. However, to date, no studies have systematically analyzed the association of distinct DM-associated autoantibodies with different histologic features routinely analyzed on a diagnostic muscle biopsy.
We identified all DM patients with banked serum and a muscle biopsy read at our institution. These sera were then screened for DM-specific autoantibodies including Mi-2, TIF1-γ, NXP2, and MDA5. We also screened sera for Jo1 autoantibodies, which are found in both DM and PM. In addition, we screened for Ro52 and PM-Scl, which, although sometimes associated with DM and PM, are not specific for patients with myositis. We then compared muscle biopsy features in patients with DM with different autoantibodies to determine whether unique histologic abnormalities are associated with different serological subtypes. In addition, because anti-Jo1–positive patients may have either DM or PM8, we also evaluated whether muscle biopsy features varied in these patients depending on whether they had a DM rash.
MATERIAL AND METHODS
Patient population
Patients seen at the Johns Hopkins Myositis Center between 2006 and 2013 were included in our study if they had (1) a Bohan and Peter diagnosis of probable or definite DM1; (2) a muscle biopsy evaluated for clinical purposes at the Johns Hopkins Neuromuscular Pathology Laboratory; and (3) banked serum to test for autoantibodies. Of note, only patients with unambiguous Gottron papules, Gottron sign, and/or heliotrope rash observed by ALM or LC-S were included; patients with only self-reported rashes were not included. Patients with Bohan and Peter probable or definite PM who were positive for anti-Jo1 and had a muscle biopsy read at Johns Hopkins were also included.
The study was approved by the Johns Hopkins institutional review board and all participants provided informed consent.
Antibody assays
All DM serum samples were tested for the most common myositis-specific (anti-Jo1, anti–Mi-2, anti-TIF1-γ, anti-MDA5, and anti-NXP2) and myositis-associated (anti-Ro52 and anti-PM-Scl) antibodies. Autoantibody testing was performed specifically for our study in batches using the same methods during 2013 and 2014. Testing for autoantibodies only rarely found in DM (e.g., non-Jo1 antisynthetase antibodies) was not undertaken. Ro52 and Jo1 antibodies were determined using commercially available ELISA kits (Inova Diagnostics). MDA5, NXP2, Mi-2, and PM-Scl antibodies were assayed by immunoprecipitation using 35S-methionine labeled proteins generated by in vitro transcription and translation (IVTT) from the appropriate cDNA, as described9. For PM-Scl, cDNA encoding the 100 and 75 kDa subunits were used to generate radiolabeled proteins, and both products were used in the immunoprecipitations to assess anti–PM-Scl antibodies. All IVTT immunoprecipitates were electrophoresed on sodium dodecyl sulfate-polyacrylamide gels and detected by fluorography. TIF1-γ antibodies were assessed by immunoprecipitation using lysates made from cells transiently transfected with TIF1-γ cDNA, followed by detection by immunoblotting as described9.
Muscle biopsies
Muscle biopsies were prospectively interpreted as part of routine clinical care at the Johns Hopkins Neuromuscular Pathology Laboratory by a histopathologist who was blinded to autoantibody status and consistently reported on the presence or absence of perifascicular atrophy, cytochrome oxidase (COX) fibers, perivascular inflammation, primary inflammation (invasion of non-necrotic fibers by mononuclear cells), and necrosis/degeneration. A single histopathologist (AMC) read 77 of 91 available biopsies (85%). To determine the interrater reliability between AMC and the other histopathologists, we compared the readings of each using 16 random DM cases from those included in this study for each of the 5 features analyzed. We found that there was excellent interrater agreement (κ = 0.81). The muscle biopsy reports were retrospectively reviewed for muscle biopsy features. Electron microscopic features and specialized immunostainings were not included in the analysis. To classify patients according to the Bohan and Peter criteria, muscle biopsies were considered compatible with an inflammatory myopathy if they showed degeneration, necrosis, myophagocytosis, and/or mononuclear cell infiltrates. Muscle biopsies were defined as revealing a necrotizing myopathy if they included necrotic myofibers (without a predominant perifascicular distribution) in the absence of perifascicular atrophy or significant endomysial or perimysial inflammation (including any primary inflammation). A subset of biopsies was stained with COX-1 and succinic dehydrogenase and mitochondrial dysfunction was defined as the presence of more than 5 COX-negative fibers per frozen section. Time from the onset of symptoms to the muscle biopsy, location of the muscle biopsy, and information about immunosuppressant treatment prior to and during muscle biopsy were recorded as potential sources of bias.
Statistical analysis
Qualitative variables were expressed as percentages and absolute frequencies while quantitative variables were expressed as median, first, and third quartiles. Fisher’s exact test and Wilcoxon rank-sum test were used to compare categorical and quantitative biopsy findings between different antibodies or antibody combinations in patients with DM and between anti-Jo1–positive patients with DM and PM.
A Poisson regression study with robust variance estimates was performed to assess the influence of treatment, time from the onset of the disease to the biopsy, and biopsy location (predictor variables) over the prevalence of the 5 biopsy features analyzed (perifascicular atrophy, perivascular inflammation, primary inflammation, predominantly necrotizing, and mitochondrial dysfunction), reporting the prevalence ratio (PR), 95% CI, and p value of significant results13.
Because this was an exploratory study, a 2-sided p value of 0.05 or less was considered significant for these analyses, with no correction for multiple comparisons.
Microsoft Access 2007 was used to do the data collection, and the statistical analyses were performed using Stata/SE 12.1 and SPSS 20.
RESULTS
Antibody assays
The commercial assays for anti-Ro52 and anti-Jo1 have been described and validated14. Our assays for anti-NXP-2, anti-TIF1-γ, and anti-MDA-5 have been described in detail and validated elsewhere9. As part of the current study, we screened 34 healthy control sera using the anti–Mi-2 and anti-PM-Scl assays; none of these tested positive.
Patients
Our study included 91 adults with DM (58 females, 82% Bohan and Peter definite DM) who had sera available and a muscle biopsy read at Johns Hopkins. Seven anti-Jo1–positive patients with PM (6 females, 43% Bohan and Peter definite PM) who had a muscle biopsy read at Johns Hopkins were also included (Table 1; Supplementary Table 1, available from the authors on request). More than half of the patients with DM were receiving treatment before (61%) and at the time of (56%) biopsy (Table 2). Most of the patients receiving immunosuppressants at the time of the biopsy were treated with corticosteroids (86% of the patients treated at biopsy; Table 2), while the next most common immunosuppressant at the time of biopsy was methotrexate (14% of the patients treated at biopsy).
Characteristics of the study subjects.
Muscle biopsy features, treatments, and duration of disease at biopsy according to autoantibody subsets in patients with dermatomyositis (DM).
Antibodies in DM
At least 1 antibody was detected in 76 patients (84%; Table 2). The most frequently detected antibodies were anti-TIF1-γ (n = 25, 27%), anti-Ro52 (n = 22, 24%), anti-NXP2 (n = 17, 19%), and anti-Jo1 (n = 13, 14%). Around one-third of patients had more than 1 antibody specificity (n = 27, 30%). Anti-Jo1 and anti-Ro52 were the 2 autoantibodies most frequently found in the same patient (n = 9, 10% of the total sample, 69% of anti-Jo1 patients; Table 2).
In 15 patients (16%), none of the antibodies systematically screened for were detected. In some of these, other autoantibodies were detected either in our laboratory or in commercial laboratories. For example, 1 patient was positive for anti-PL-12 and 1 patient was anti-U1RNP–positive. Among the remaining 13 patients, immunoprecipitations from radioactively labeled HeLa cells revealed numerous unidentified bands, but no patterns were shared between the different patients (data not shown). Because these patients were unlikely to represent a serologically homogeneous group, they were excluded from subgroup analyses.
Muscle biopsy features in DM
Perivascular inflammation (n = 56, 62%) and perifascicular atrophy (n = 46, 51%) were the most frequent muscle biopsy findings in patients with DM (Table 2). Other less commonly observed features of DM included primary inflammation (n = 21, 23%) and mitochondrial dysfunction, which was found in 14 of 50 biopsies (28%) stained with COX. A predominantly necrotizing myopathy was observed in some patients with DM (n = 15, 16%).
Regression analysis showed that the prevalence of each muscle biopsy feature was not significantly associated with the time from the onset of the disease to the biopsy. However, patients without immunosuppressant treatment during biopsy had an increased prevalence of perivascular inflammation (PR 1.38, 95% CI 1.00–1.90, p = 0.05). Biopsies taken from deltoid muscle, compared to those taken from quadriceps, showed significantly more perivascular inflammation (PR 2.16, 95% CI 1.44–3.24, p < 0.001), perifascicular atrophy (PR 1.88, 95% CI 1.11–3.18, p = 0.02), and mitochondrial dysfunction (PR 4.25, 95% CI 1.02–17.80, p = 0.05). Primary inflammation and necrotizing muscle biopsy findings were not significantly influenced by these potential confounders.
Correlating muscle biopsy features with autoantibody status in DM
We compared muscle biopsy features of all patients positive for each antibody with all those negative for the same antibody (Table 3). Univariate analysis revealed that TIF1-γ+ patients had more mitochondrial dysfunction than did TIF1-γ– patients (47% vs 18%; p = 0.05). NXP2+ patients had less primary inflammation than those without this autoantibody (0% vs 28%; p = 0.01). In contrast, primary inflammation was more common in Mi-2+ (50% vs 19%; p = 0.03) and PM-Scl+ patients (67% vs 18%; p = 0.004) compared to those without the respective autoantibodies. Although reliability was limited owing to small sample numbers, multivariate analysis confirmed that TIF1-γ+ patients had more mitochondrial dysfunction (PR 2.6, 95% CI 1.0–6.5; p = 0.05) and PM-Scl+ patients had more primary inflammation (PR 5.2, 95% CI 2.0–13.4; p = 0.001) independent of disease duration at biopsy, biopsy site, and treatment at biopsy. There was excellent interrater agreement among the histopathologists who interpreted these biopsies (κ = 0.93). However, we also performed univariate and multivariate analyses using only the 77 biopsies read by AMC, and the associations described above were preserved.
Histological features, treatment, and biopsy site characteristics of dermatomyositis (DM) patients with and without individual DM autoantibodies. Numbers are percentages unless otherwise indicated.
We also compared the prevalence of muscle biopsy features of each autoantibody subgroup against each of the other autoantibody subgroups. Statistically significant differences in the prevalence of primary inflammation were found between anti–Mi-2 and anti-TIF1-γ (p = 0.005), anti–Mi-2 and anti-NXP2 (p = 0.002), anti-PM-Scl and anti-TIF1-γ (p = 0.004), anti-PM-Scl and anti-NXP2 (p < 0.001), anti-NXP2 and anti-Ro52 (p = 0.03), anti-PM-Scl and anti-MDA5 (p = 0.03), and anti-NXP2 and anti-Jo1 (p = 0.03). Compared with biopsies from patients positive for both anti-Jo1 and anti-Ro52, biopsies from patients positive for anti-Jo1 alone showed significantly less perivascular inflammation (p = 0.05). Statistically significant differences in the prevalence of perivascular inflammation were also found between anti-MDA5 and anti–Mi-2 (p = 0.03).
Given that immunosuppressive treatment could affect muscle biopsy findings, we compared the prevalence of such treatment before biopsy and at the time of biopsy between each autoantibody subgroup. Anti-NXP2 patients were less frequently treated at biopsy than anti-Ro52 (p = 0.03) subjects and less frequently before biopsy than anti-MDA5 subjects (p = 0.05). The time between symptom onset and muscle biopsy was also evaluated between each antibody subgroup. The time from symptom onset to biopsy was longer in anti-MDA5+ subjects compared to those with anti-NXP2 (p = 0.03) and in those with anti-Jo1 compared to those with anti–Mi-2 (p = 0.02) or anti-NXP2 (p = 0.003). The interval between symptom onset and muscle biopsy was also longer in those with anti-Ro52 compared to those with either anti-NXP2 (p = 0.003), anti-TIF1 (p = 0.04), or anti–Mi-2 biopsies (p = 0.01). However, with the exception of comparisons with anti-MDA5–positive patients, who had long disease duration and were all treated at the time of biopsy, differences in treatment or disease duration at biopsy did not appear to account for differences in muscle biopsy features among the different autoantibody subgroups. Multivariate analyses to explore the role of potential confounders was not possible given the small size of individual autoantibody subgroups.
Comparison of muscle biopsy features between DM and PM in anti-Jo1–positive patients
There were no significant differences in muscle biopsy features between DM and PM patients who were positive for anti-Jo1 (Table 4). Indeed, 4 of 7 patients (57.1%) with PM had perifascicular atrophy, considered the hallmark of DM, and 4 of 13 patients (31%) with DM had primary inflammation. Of note, there were no significant differences in treatment or duration of symptoms at the time of biopsy between anti-Jo1 DM and PM patients.
Muscle biopsy features, treatments, and duration of disease in anti-Jo1–positive patients diagnosed with DM and PM.
DISCUSSION
This is the first study, to our knowledge, to systematically compare muscle biopsies from patients with DM who had different autoantibodies. Certain features were relatively common among most patients with DM regardless of autoantibody specificity. These included perivascular inflammation and perifascicular atrophy, which were found in 62% and 51% of all patients with DM, respectively. In contrast, mitochondrial dysfunction, a described feature in DM muscle biopsies6, was relatively rare (28%) except in those with anti-TIF1-γ, where it was found in 47% of patients. However, the prevalence of other muscle biopsy features varied significantly depending upon the autoantibody status. Most strikingly, primary inflammation was present in the majority of 21 patients with antibodies against either Mi-2 (50%) or PM-Scl (67%) but not in any of the 17 anti-NXP2 patients.
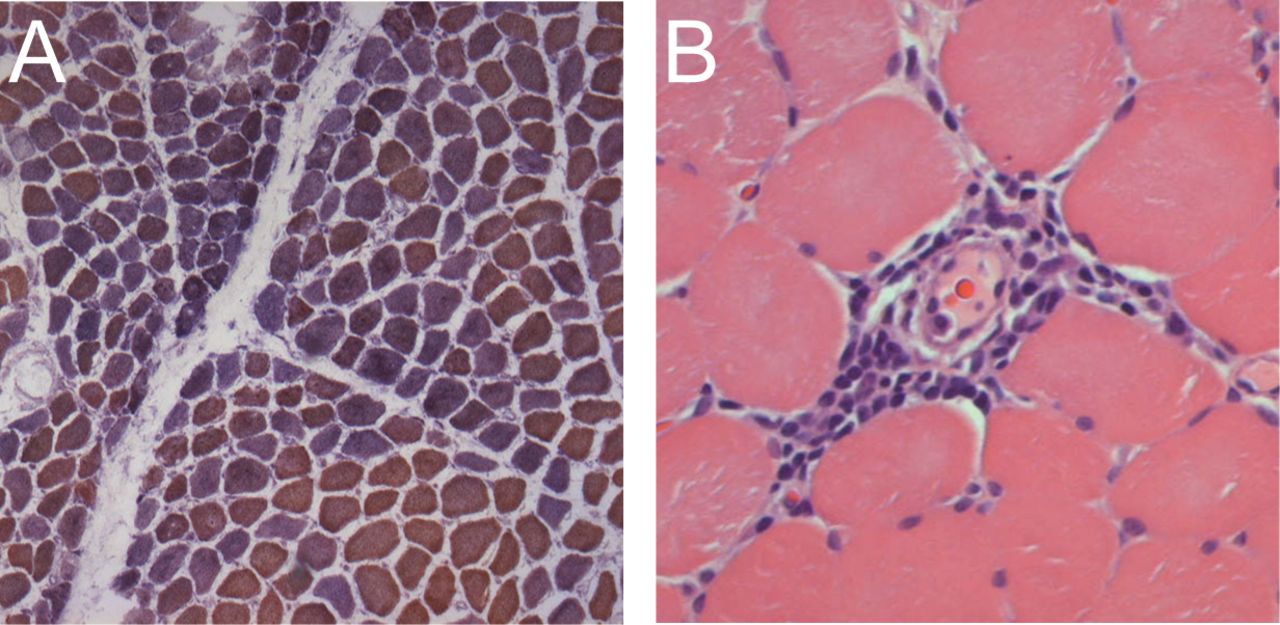
Our study reveals for the first time, to our knowledge, that patients with anti-TIF1-γ and anti-NXP-2 have very similar histologic profiles, with prominent perifascicular atrophy and perivascular inflammation but very little primary inflammation. The main difference between muscle biopsies in patients with these 2 serologies is that only anti-TIF1-γ patients have a relatively high prevalence of mitochondrial dysfunction. Figure 1 shows an example of a typical TIF1-γ muscle biopsy with perifascicular atrophy, perivascular inflammation, and mitochondrial dysfunction.

Typical muscle biopsy from patient with DM who is positive for transcriptional intermediary factor 1-γ. A. Low-power view of a frozen section stained with cytochrome oxidase (COX; brown) and succinic dehydrogenase (blue) reveals both normal fibers (brown) and numerous COX-deficient fibers (purple/blue) indicating mitochondrial dysfunction. Several fascicles include examples of perifascicular atrophy. B. This high-power view of a paraffin section from the same patient stained with H&E shows a striking example of perivascular inflammation.
Patients with anti–Mi-2 antibodies had the highest prevalence of perifascicular atrophy (67%) and perivascular inflammation (83%) compared to those with one of the other DM-specific autoantibodies considered individually (although this was not statistically significant). They also had a higher prevalence of primary inflammation (51%) compared to those without this DM-specific autoantibody, although anti-PM-Scl–positive patients had an even higher prevalence (67%) of this pathologic feature. Of note, patients with anti–Mi-2 are known to have other distinctive clinical features including a severe rash that responds well to immunosuppression and a low cancer rate15.
Although previously investigated in juvenile patients with DM16, our study includes the first description, to our knowledge, of muscle biopsies from adult anti-MDA5–positive patients with DM. Despite the fact that anti-MDA5 has been linked with clinically amyopathic DM12, we had 5 biopsies available from patients with this immunospecificity who had 2 or more myopathic features required for a diagnosis of probable or definite DM by Bohan and Peter criteria (i.e., proximal muscle weakness, elevated muscle enzymes, irritable myopathy on electromyography, and/or characteristic muscle biopsy features). Compared to other adult patients with DM, these anti-MDA5–positive subjects had a low prevalence of the histologic features analyzed in this study, including perivascular inflammation, primary inflammation, and perifascicular atrophy. Scattered atrophic fibers were the only histologic abnormality noted on 3 of 5 biopsies from the anti-MDA5–positive subjects. We speculate that the high frequency of treatment at the time of biopsy (100%) may account for the relatively bland biopsies. Indeed, our regression analysis indicated that perivascular inflammation is less prevalent when patients are treated before muscle biopsy.
About one-third of patients were positive for more than 1 of the autoantibodies we screened for, especially the combination of anti-Jo1 and anti-Ro52. It is well established that the combination of these antibodies may be associated with severe myositis and joint impairment17,18. Thus, it is of interest that in the current study we found a significantly higher prevalence of perivascular inflammation in anti-Jo1 with anti-Ro52 compared with anti-Jo1 alone (89% vs 25%, p = 0.05), suggesting a more intense inflammatory phenomenon in the former group of patients.
In addition to comparing the muscle biopsy features in patients with DM with different autoantibodies, we also compared the muscle biopsies of anti-Jo1–positive patients who were diagnosed with either DM or PM based on Bohan and Peter criteria. Surprisingly, there were no significant differences in the basic histologic features between anti-Jo1–positive patients with and without a typical DM rash. Indeed, 4 of 7 (57%) anti-Jo1–positive patients without rash were found to have perifascicular atrophy, considered a histologic hallmark of DM2. Based on these findings and a lack of data indicating that anti-Jo1–positive patients with and without rash are pathophysiologically distinct, we suggest that all anti-Jo1–positive patients have the same disease and should not be categorized as having DM or PM. Rather, we propose that the anti-Jo1 syndrome could be considered a single entity characterized by the presence of the antibody along with 2 or more of the following features of the antisynthetase syndrome: myositis, interstitial lung disease, rash, arthritis, mechanic’s hands, and Raynaud phenomenon.
Of note, our study showed that 16% of patients with Bohan and Peter probable or definite DM did not have perifascicular atrophy, primary inflammation, or perivascular inflammation on muscle biopsy. Rather, almost 1 in 6 patients with DM have a necrotizing muscle biopsy that cannot readily be distinguished from patients with IMNM/NAM and either anti-SRP or anti-HMGCR autoantibodies7. Therefore, in clinical practice, it may be that only autoantibody testing can reliably distinguish between a patient with DM sine dermatitis and IMNM/NAM.
Interestingly, Poisson regression analysis showed that biopsies from the deltoid muscle and those taken during periods without immunosuppressant treatment were more likely to show perivascular inflammation and perifascicular atrophy. This suggests that the diagnostic performance of the muscle biopsy may be influenced by both treatment and muscle biopsy location.
Our study has a number of limitations. First, given the rarity of DM, the number of patients studied in some antibody groups was small (for example, MDA5 and PM-Scl) and consequently the study may have been underpowered to detect all clinically relevant associations. Second, except for anti-Jo1 patients with and without anti-Ro52, we did not have adequate numbers of patients with the same combination of multiple antibodies to study these as distinct groups. Third, although all muscle biopsies were interpreted at Johns Hopkins, the biopsies were performed at various institutions, biopsy location was highly variable, and muscle tissue was not available for further study for a majority of the study subjects. Therefore, we used only those features that were assessed for clinical purposes through routine histological methods. Fourth, muscle biopsy features were categorized as either present or absent, and so the severity of these features could not be compared between subgroups. Fifth, the analysis did not include electron microscopy or specialized immunostaining for MHC I, the membrane attack complex, or inflammatory cell subsets (e.g., CD8-positive cells). Comparing specialized immunostaining in DM patients with different autoantibodies would be of interest. Sixth, not all biopsies included in this study were stained for COX, so the data on mitochondrial dysfunction are not complete. Finally, despite the differences we have emphasized, it should be noted that even among those with a given autoantibody, there is considerable variability in the observed muscle biopsy features. Thus, an individual’s autoantibody status cannot be reliably inferred from the histologic features noted on muscle biopsy.
These limitations notwithstanding, our study provides the first comparative description of muscle biopsies from patients with DM with different autoantibodies. Further, our study demonstrates that the prevalence of different histological features varies according to autoantibody status in DM. This raises the possibility that different pathologic pathways underlie muscle disease in patients with different autoantibodies. Along with prior work showing that each autoantibody is associated with different disease manifestations (e.g., cancer), our findings further support the conclusion that DM is not a homogeneous entity, but may consist of several different diseases with distinct biomarkers (i.e., autoantibodies). Our findings also support the possibility that patients with anti-Jo1 antibodies have a single disease, the antisynthetase syndrome (rather than PM or DM), which sometimes includes rash as a prominent feature. This framework for understanding the relationship between different autoantibodies and distinct disease states remains to be validated in other cohorts.
Footnotes
The Johns Hopkins Rheumatic Disease Research Core Center, where the autoantibody assays were performed, is supported by NIH grant P30-AR-053503. Ms. Casciola-Rosen is supported by grant R56 AR062615-01A1. Dr. Christopher-Stine’s work was supported by the Huayi and Siuling Zhang Discovery Fund. This research was supported (in part) by the Intramural Research Program of the NIAMS of the NIH.
- Accepted for publication April 24, 2015.








{kind=link}